










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Obstetricia y Ginecología
Print version ISSN 0034-7434On-line version ISSN 2463-0225
Rev Colomb Obstet Ginecol vol.55 no.3 Bogotá July/Sept. 2004
* Residente de Tercer Año Hospital Militar Central. Departamento de Ginecología y Obstetricia, Facultad de Medicina. Universidad Militar Nueva Granada, Bogotá D.C., Colombia. Correo electrónico: alvinzy@hotmail.com
** Especialista en Ginecología y Obstetricia. Jefe de Medicina Materno Fetal. Jefe de la Unidad de Ultrasonido Doppler. Fellow Unidad de Cuidados Intensivos. Profesor Titular de Ginecología y Obstetricia. Hospital Militar Central, Facultad de Medicina. Universidad Militar Nueva Granada. Profesor Titular de la Unidad de Medicina Materno Fetal, Hospital Universitario La Samaritana.
*** Bacterióloga Universidad Colegio Mayor de Cundinamarca. Especialista en Epidemiología, Universidad Colegio Mayor del Rosario. Bacterióloga Servicio de Hematología Especial, Coordinadora del Laboratorio de Coagulación del Hospital Militar Central.
RESUMEN
El síndrome de la plaqueta pegajosa es un desorden plaquetario autosómico dominante asociado con eventos tromboembólicos arteriales y venosos. Se caracteriza por hiperagregabilidad plaquetaria en plasma rico en adenosindifosfato y epinefrina. Clínicamente las pacientes pueden presentar angina de pecho, infarto agudo del miocardio, accidentes isquémicos transitorios, accidentes cerebrovasculares, trombosis retiniana, trombosis de arterias periféricas y trombosis venosas, frecuentemente recurrente bajo tratamiento anticoagulante oral. Se presentan cuatro casos de gestantes diagnosticadas y tratadas en la Unidad de Alto Riesgo Obstétrico del Hospital Militar Central, con pruebas diagnósticas para otros eventos hipercoagulables normales que sólo presentaban hiperagregación plaquetaria. Posteriormente se hace una revisión de la literatura.
Palabras clave: desorden plaquetario, agregación celular.
SUMMARY
The sticky platelet syndrome is an autosomic dominant disorder of the platelets with arterial and venous thromboembolic events. The syndrome characterizes by hyper aggregation of platelet in the plasma enriched in adenosindiphosphate and epinephrine. Clinically, the patients can present or display chest angina, acute infarct of the myocardium, transitory cerebro-vascular ischemic accidents, retinal, periphery arteries, and venous thrombotic accidents, frequently recurrent even under oral anticoagulation treatment. We present 4 cases of pregnant women seen at the high risk obstetric Unit at the Central Military Hospital in Bogotá, Colombia, with normal coagulation tests and that only displayed hyper aggregation of the platelets. A review of literature is presented.
Key words: platelet disorder, cell aggregations.
CASO CLÍNICO 1
Paciente de 25 años, que consulta por cuadro clínico de ocho meses de evolución de dolor en miembro inferior izquierdo, con edema y claudicación intermitente.
Antecedentes personales: trombosis venosa profunda en anterior gestación desde la semana 4, no recuerda manejo médico en el Hospital General de Barranquilla, Colombia.
Antecedentes ginecoobstétricos: menarquia 12 años, ciclos regulares. Gestante 2, partos 1, cesárea 1 (hace cuatro años en Hospital General de Barranquilla por feto macrosómico). Fecha de última menstruación: diciembre 17 de 2002.
Al examen físico presentó como datos positivos: aumento del diámetro de pierna izquierda de 2 cm en relación con la derecha. Doppler venoso (febrero 2 de 2003): cambios sugestivos de trombosis venosa profunda subaguda y crónica de vena poplítea izquierda.
Se inicia manejo con enoxaparina 60 mg subcutáneos/día desde la semana 10 de gestación, sin mejoría clínica. Consulta en varias ocasiones a la urgencia de la institución; valorada por Cirugía Vascular y Grupo de Alto Riesgo Obstétrico, quienes le realizan paraclínicos y se decide continuar con igual manejo.
Exámenes paraclínicos: Doppler venoso de miembros inferiores (agosto 8 de 2003): trombos mixtos en venas poplíteas, gemelares y tronco tibio peroneo; mal recanalizadas, con pobre incremento del flujo. Trombosis venosa intrapatelar con compromiso de la vena tibial posterior en su segmento distal.
Anticuerpos antifosfolípidos: inmunoglobulina G y M negativos. Anticoagulante lúpico: negativo. Proteína C y S: negativas. Resistencia a la proteína C: negativa.
Curvas de agregación plaquetaria:
Agregación con epinefrina a diferentes concentraciones: 11 uM/ml: 98,2% (VN: 39-87%); 1,1 uM/ml: 91,8% (VN: 15-55%); 0,6 uM/ml: 21,4% (VN: 0-35%).
Agregación con adenosindifosfato (ADP) a diferentes concentraciones: 2,3 uM/ml: 97,7% (VN: 28-75%); 1,2 uM/ml: 99% (VN: 15-51%); 0,6 uM/ml: 21,4% (VN: 10-36%) (gráfica 1).
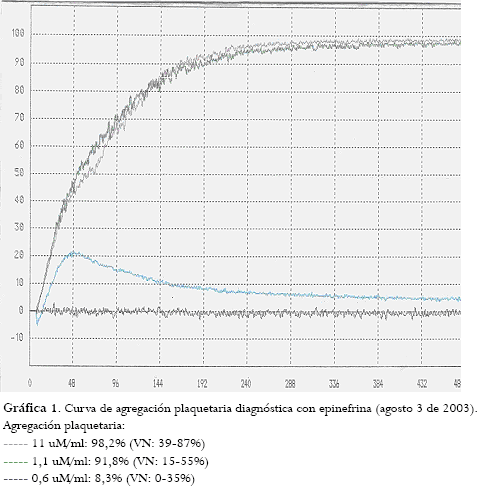
El día del estudio completo para diagnóstico de síndrome de plaqueta pegajosa (SPP) se le inició manejo con ácido acetilsalicílico (ASA), 100 mg/VO/día. Debido a la ausencia de respuesta clínica y a la curva de agregación plaquetaria alterada a las dos semanas de tratamiento, se aumentó la dosis a 200 mg/VO/día con mejoría clínica y curva de agregación plaquetaria de control normal.
Se suspende el tratamiento a la semana 36 de gestación. A las 38 semanas se realiza cesárea por RPM sin trabajo de parto y antecedente de cesárea.
CASO CLÍNICO 2
Paciente de 28 años. Acude a consulta de alto riesgo por antecedente de pérdida fetal, cesárea previa y cefalea que no cede con la administración de acetaminofén. Refiere que se le inició tratamiento con ASA al inicio de la gestación actual sin conocer los motivos.
Antecedentes personales: migraña, tratamiento con nimesulide con leve mejoría y actualmente con acetaminofén, sin mejoría. Agenesia renal izquierda. Refiere que durante la gestación con embarazo a término recibió tratamiento con ASA, 100 mg/día; no especifica causa.
Antecedentes familiares: madre con migraña.
Antecedentes ginecoobstétricos: menarquia a los 12 años, ciclos 28/3. Gastante 3, partos 1, cesárea 1 (8 años), aborto 1 (agosto 21 de 2001) a la semana 7 presenta aborto retenido. Fecha de la última menstruación: agosto 24 de 2002.
Exámenes paraclínicos: anticuerpos antifosfolípidos: negativos. Anticoagulante lúpico: negativo. Proteína C y S: negativas. Resistencia a la proteína C: negativa.
Curvas de agregación plaquetaria:
Agregación con epinefrina a diferentes concentraciones: 11 uM/ml: 85% (VN: 39-87%), 1,1uM/ml: 14,1% (VN: 15-55%), 0,6 uM/ml: 8,2% (VN: 0-35%).
Agregación con ADP a diferentes concentraciones: 2,3 uM/ml: 86,4% (VN: 28-75%), 1,2 uM/ml: 83,2% (VN: 15-51%), 0,6 uM/ml: 33,2% (VN: 10-36%).
Con diagnóstico de SPP se inicia tratamiento con ASA, 100 mg/VO/día, con mejoría del cuadro clínico y curva de control de agregación plaquetaria a los 15 días de tratamiento normal.
CASO CLÍNICO 3
Paciente de 35 años, acude a Consulta de Alto Riesgo por dolor y edema en miembros inferiores.
Antecedentes patológicos: migraña y trombosis venosa profunda que no mejoró con enoxaparina.
Antecedentes familiares: cardiopatía, IAM y migraña.
Antecedentes ginecoobstétricos: menarquia a los 12 años, ciclos 28/3, gestante 5, partos 4, ectópico 1, vivo 1. Primer embarazo: gemelar en 1989, parto vaginal, los dos fetos fallecieron por membrana hialina; segundo parto: eutósico en 1994 con antecedente de amenaza de parto pretérmino; tercero parto: en 1998, falleció por abruptio en el expulsivo; cuarto embarazo: ectópico.
Exámenes paraclínicos: anticuerpos antifosfolípidos: negativos. Anticoagulante lúpico: negativo. Proteína C y S: negativas. Resistencia a la proteína C: negativa. Antitrombina 3: negativa.
Curva de agregación plaquetaria:
Agregación con epinefrina a diferentes concentraciones: 11 uM/ml: 78% (VN: 39-87%), 1,1uM/ml: 83% (VN: 15-55%), 0,6 uM/ml: 80,9% (VN: 0-35%).
Agregación con ADP a diferentes concentraciones: 2,3 uM/ml: 84% (VN: 28-75%), 1,2 uM/ml: 83,2% (VN: 15-51%), 0,6 uM/ml: 83,6% (VN: 10-36%).
Se inicia manejo con ASA desde la semana 29 de gestación, 100 mg/VO/día, con mejoría de la cefalea y dolor en miembros inferiores y curva de agregación plaquetaria de control normal.
CASO CLÍNICO 4
Paciente de 32 años, acude a Consulta de Infertilidad con diagnóstico de pérdida recurrente del embarazo.
Antecedentes patológicos: migraña, trombosis venosa profunda que no mejora con tratamiento con enoxaparina.
Antecedentes familiares: madre con migraña y accidente cerebrovascular. Padre con IAM.
Antecedentes quirúrgicos: laparo-histeroscopia normal.
Antecedentes ginecoobstétricos: menarquia a los 12 años, ciclos 28/4-5. Gestante 4, partos 0, abortos 4. Los cuatro abortos ocurrieron entre la semana 4 y 6 de gestación.
Exámenes paraclínicos: anticuerpos antifosfolípidos: negativos. Anticoagulante lúpico: negativo. Proteína C y S: negativas. Resistencia a la proteína C: negativa. Antitrombina 3: negativa.
Curva de agregación plaquetaria:
Agregación con epinefrina a diferentes concentraciones: 11 uM/ml: 78,2% (VN: 39-87%), 1,1 uM/ml: 88,2% (VN: 15-55%), 0,6 uM/ml: 81,8% (VN: 0-35%.
Agregación ADP a diferentes concentraciones: 2,3 uM/ml: 84,5% (VN: 28-75%), 1,2 uM/ml: 15% (VN: 15-51%), 0,6 uM/ml: 1,4% (VN: 10-36%).
Se inicia tratamiento desde la semana 5 de gestación con ASA, 100 mg/día, sin reversión de la curva de agregación plaquetaría ni mejoría de la clínica de trombosis venosa profunda ni de la migraña. Se aumenta la dosis de ASA a 200 mg/día, con reversión a la normalidad de la curva de agregación plaquetaria y mejoría de los síntomas de trombosis venosa profunda y de la migraña.
REVISIÓN DE LA LITERATURA
Definición
El síndrome de la plaqueta pegajosa (SPP) es un desorden plaquetario autosómico dominante, considerado como una de las causas más frecuentes de eventos trombóticos (tanto arteriales como venosos), a pesar de ser para muchos una patología desconocida. Desde hace algún tiempo se le conoce como la segunda causa dentro de los trastornos hereditarios relacionados con los problemas trombóticos, después de la resistencia a la proteína C activada y parece corresponder a la principal causa de trombosis arteriales. Se cree que el defecto específico puede estar localizado en los receptores de la superficie plaquetaria y se caracteriza por hiperagregabilidad de las plaquetas con ADP y/o epinefrina. El SPP es un fenómeno de hipercoagulabilidad que cada vez se identifica con mayor frecuencia, en la medida en que se han conocido sus factores asociados.¹
En la actualidad se sabe de muchos eventos tromboembólicos asociados con deficiencias de factores de anticoagulación, defectos del sistema fibrinolítico, a eventos autoinmunes, trastornos metabólicos, catéteres, enfermedades previas como el síndrome nefrótico, las hepatopatías y las enfermedades oncológicas entre otras.2
Historia
Los defectos plaquetarios han sido considerados como posibles inductores de fenómenos de hipercoagulabilidad, pero se ha creído que se trata de patologías muy raras. En 1979, Mefty A.L. y colaboradores describieron un grupo de pacientes adultos con cuadros de accidentes isquémicos transitorios, en quienes no se habían encontrado factores asociados, excepto agregación y adhesividad plaquetaria aumentadas.3
En 1983, Holliday y Mammen, en la IX Conferencia Internacional sobre Trombosis y Circulación General en Phoenix, Arizona, describieron un síndrome relacionado con infarto cerebral en adultos jóvenes y lo describieron como "síndrome de la plaqueta pegajosa".4
En 1986, Melvyn y Mammen y colaboradores presentaron un estudio realizado en 41 pacientes adultos con dolor precordial y arterias coronarias angiográficamente normales, en quienes encontraron hiperagregabilidad plaquetaria en pruebas realizadas con ADP y epinefrina, las cuales resultaron estadísticamente significativas (p<0,001) en comparación con la realizada en 73 controles.4-5
En 1995, Mammen publicó un estudio de diez años de experiencia con el SPP, identificado en más de 200 pacientes jóvenes entre 5 y 45 años y sus familias, quienes presentaban trombosis principalmente arteriales y venosas en varios casos, sin factores asociados. Todos mostraban hiperagregabilidad con ADP y/o epinefrina.6
En los primeros trabajos de Mammen se utilizaron pruebas de microscopía electrónica para poner en evidencia la respuesta de las plaquetas a una superficie de contacto (adhesividad), según la técnica descrita por Barnhart y colaboradores.
Con base en esta técnica, en un recuento de 100 plaquetas se clasifican las formas en:
Redonda o con mínima modificación en la forma (abortiva) como inactiva.
Forma dendrítica como activación intermedia.
Alteración extendida y diseminada como máxima activación.
El número de plaquetas que se agregan unas a otras también se cuenta y se expresa en porcentaje.
En los pacientes con SPP usualmente se encuentran hasta 10% de formas redondas o abortivas, 80% de formas dendríticas y 10% de formas diseminadas o extendidas, hasta con un 25% de agregados plaquetarios. En la actualidad esta técnica no se utiliza y el diagnóstico se hace por agregometría.7
En 1997, el doctor Rodger Bick publica un estudio realizado en 153 pacientes adultos con cuadros trombóticos referidos al centro clínico de trombosis y hemostasis de Dallas durante un periodo de 2 años, encontrando que del total de trombosis venosas, un 14% correspondieron al síndrome de la plaqueta pegajosa y de los eventos arteriales, un 23% correspondían a esta patología. 8
Clínica
Clínicamente la gestante presenta fenómenos de trombosis periféricas arteriales o venosas, trombosis retiniana y síndrome de muerte fetal intrauterina.9
La agregación plaquetaria se ha relacionado con los ciclos circadianos, encontrando un predominio de aparición en las horas de la mañana.10
También se ha descrito que este síndrome se puede asociar con otras anormalidades congénitas de los mecanismos de anticoagulación, como resistencia a la proteína C activada y deficiencia de proteína S.
Se han descrito muchas patologías que cursan con hiperagregabilidad plaquetaria, como diabetes mellitus, síndrome nefrítico, fibrosis quística y anorexia nerviosa, entre otras. En estos casos se ha encontrado que los niveles del factor 4 plaquetario (PF4), beta-trombo globulina y tromboxano A2 se encuentran elevados, lo cual sugiere la existencia de mecanismos intrínsecos, probablemente secundarios a activación plaquetaria en la circulación.11
También es importante tener en cuenta que los defectos congénitos de la lipooxigenasa generan trombosis sin elevación del FP4 ni de la beta-trombo globulina.12
Diagnóstico
El diagnóstico se fundamenta en la demostración de la hiperagregabilidad mediante pruebas de agregometría. Sin embargo, se sabe que estas pruebas se han diseñado especialmente para la demostración de los defectos en la agregación y no para la respuesta plaquetaria aumentada. Además, son conocidas las dificultades de estandarización de las pruebas.12
Uno de los sistemas más recientes de análisis de la función plaquetaria, el PFA-100, se ha considerado como uno de los más seguros.12
En todos los pacientes con SPP, las pruebas de agregación con colágeno, ácido araquidónico y ristocetina son normales.
Las pruebas con epinefrina se realizan con plasma rico en plaquetas y epinefrina a concentraciones de 11, 1,1 y 0,6 uM/ml.
Las pruebas con ADP se realizan con concentraciones de 2,3, 1,2 y 0,6 uM/ml.
La agregación se expresa en porcentaje de transmisión de la luz; 100% es agregación completa y 0% ausencia de agregación.
En los pacientes normales, al diluir la epinefrina y el ADP, las curvas muestran disminución de la agregación, mientras que en el paciente que presenta el SPP, las curvas de la agregación permanecen elevadas.
Se deben descartar las patologías ya descritas que aumenten la agregación plaquetaria.
En los cuadros agudos de trombosis arteriales o venosas se han encontrado fenómenos de hiperagregabilidad, los cuales podrían ser consecuencia de activación del sistema hemostático. Por esta razón, las pruebas mencionadas se deben realizar o repetir cuatro a nueve meses después del evento.
Criterios diagnósticos
Se consideran tres tipos diferentes del SPP:
Tipo I: hiperagregabilidad tanto con epinefrina como con ADP.
Tipo II: hiperagregabilidad solamente con epinefrina.
Tipo III: Hiperagregabilidad solamente con ADP.
Diagnóstico sugestivo:
Hiperagregabilidad con sólo una concentración de un reactivo e historia de trombosis. El diagnóstico se confirma si al repetir la prueba se corrobora la misma anormalidad.
Diagnóstico confirmado:
Historia de trombosis e hiperagregabilidad con dos concentraciones de un reactivo, 0.
Historia de trombosis e hiperagregabilidad con una concentración de ambos reactivos, 0.
Historia de trombosis e hiperagregabilidad con una concentración de un solo reactivo, si al repetir la prueba se confirma el mismo resultado.
Una circunstancia de mucho valor es la hiperagregabilidad en las pruebas de agregación de los padres, hermanos o familiares asintomáticos o con antecedentes de trombosis.
En el SPP, las pruebas de PF4 y beta-tromboglobulina son siempre negativas y el tiempo de sangría permanece anormal.
El SPP se debe sospechar en pacientes en quienes a pesar de estar bajo anticoagulación oral presentan recurrencias del evento trombótico.
Las pruebas deben realizarse por lo menos dos semanas después de haber suspendido cualquier medicamento que altere la función plaquetaria.
Usualmente los familiares cercanos han tenido antecedentes trombóticos y con frecuencia se encuentran antecedentes de migraña.13
Recomendaciones
Las pruebas se deben realizar en más de una oportunidad, con resultados similares.
Los hallazgos de laboratorio deben desaparecer durante el tratamiento con ASA.
Los hallazgos deben reaparecer cuando se repitan las pruebas al suspender el ASA por lo menos con 15 días de anticipación.
Se debe establecer un patrón familiar mediante pruebas realizadas a padres, hermanos y otros familiares.
Se deben realizar todas las demás pruebas de hipercoagulabilidad, por lo menos las más frecuentemente descritas.
Si las pruebas se han efectuado durante el evento agudo, se deben repetir luego de cuatro a nueve meses después de suspendido el ASA por lo menos 15 días antes.
Tratamiento
ASA
Se utilizan dosis de 80 mg diarios con normalización del patrón hiperagregable en la inmensa mayoría de los casos y con mejoría de la sintomatología.
El efecto antiagregante plaquetario se debe a su capacidad como donante del grupo acetilo a la membrana plaquetaria y a la inhibición irreversible de la enzima ciclooxigenasa. Inhibe la actividad de la enzima ciclooxigenasa, y esto a su vez la formación de los precursores de las prostaglandinas y tromboxanos a partir del ácido araquidónico. Aunque la mayoría de sus efectos terapéuticos puede deberse a la inhibición de la síntesis de prostaglandinas en diferentes tejidos.
El ASA se opone a la agregación plaquetaria por la inhibición selectiva de la ciclooxigenasa plaquetaria, lo cual disminuye la producción del tromboxano A2 (potente vasoconstrictor y estimulador de la agregación plaquetaria) y modifica el equilibrio entre estas dos sustancias antagónicas.
En algunos casos es necesario aumentar la dosis hasta 325 mg diarios para lograr la normalización.14
El ASA no presenta riesgos para el feto. Debe suspenderse su administración en caso de presentarse amenaza de parto pretérmino y cuando se planea una cordocentesis.
También se excreta en la leche materna, observándose que tras la ingestión de una dosis única de 325 mg se han detectado concentraciones máximas de salicilato de 86-241 ug/ml.
En el último trimestre puede prolongar el trabajo de parto y contribuir al sangrado materno y fetal. En pacientes asmáticas puede precipitar una crisis.
La absorción es rápida y completa tras la administración oral; los alimentos disminuyen la velocidad pero no el grado de absorción. Su unión a las proteínas (albúmina) es alta, pero decrece según aumenta la concentración plasmática, con concentraciones bajas de albúmina, en la disfunción renal y durante el embarazo.
Puede inducir broncospasmo en pacientes con asma, alergias y pólipos nasales.
Está contraindicada en pacientes epilépticas que reciben hidantoína, en pacientes con antecedentes de abruptio y placenta previa.
Los salicilatos desplazan en su ligadura proteínica a las sulfonilureas, penicilina, tiroxina, triyodotironina, fenitoína y naproxeno y potencian sus efectos. El uso simultáneo con otros analgésicos antiinflamatorios no esteroides puede aumentar el riesgo de hemorragias debido a la inhibición aditiva de la agregación plaquetaria. Los glucocorticoides incrementan la excreción de salicilato por lo que se deberá adecuar la dosificación.
Debe evaluarse la relación riesgo-beneficio en pacientes con anemia, asma, alergias, gota, tirotoxicosis, ya que pueden exacerbarse con dosis elevadas.
Algunos anestesiólogos no recomiendan anestesia regional en caso de no haber suspendido el fármaco una semana antes.
Clorhidrato de ticlopidina
Es un antiagregante plaquetario específico. Se utilizan dosis de 250-500 mg/día.
Contraindicaciones: diátesis hemorrágica, úlcera gastroduodenal, accidente hemorrágico, hemopatías que conllevan un alargamiento del tiempo de sangrado, antecedente de leucopenia, trombocitopenia y agranulocitosis.
Debe evitarse el uso concomitante con salicilatos, AINE, anticoagulantes orales y heparinas, porque aumentan el riesgo hemorrágico.
La inocuidad de la administración de ticlopidina durante el embarazo o en el periodo de lactancia no ha sido establecida; los estudios en ratas han demostrado que se excreta por la leche materna. Salvo en los casos de indicación formal, es conveniente evitar su uso durante el embarazo o la lactancia.15
CONCLUSIÓN
El SPP, no obstante ser una de las causas más frecuentes de eventos trombóticos, tanto arteriales como venosos, es considerado por muchos como una patología desconocida. A pesar que la descripción de este síndrome se ha realizado en adultos con antecedentes de enfermedad tromboembólica, IAM y accidente cerebrovascular, su incidencia en la población gestante es desconocida. Por sus características clínicas y su relación con pérdidas gestacionales es importante tenerlo en cuenta en el diagnóstico diferencial de las trombofilias en pacientes gestantes con antecedentes de pérdidas gestacionales y de trombosis venosa profunda.
REFERENCIAS
1. Chittoor SR, Elsehety AE, Roberts GF, Laughlin WR. Sticky Platelet Syndrome: a case report and review of the literature. Clin Appl Thromb Hemost 1998;4:280-4. [ Links ]
2. Andrews M, Monagle PT, Brooker L. Congenital Prothrombic disorders: presentation during infancy and childhood in thromboembolic complications during infancy and childhood. BC Decker Inc.; 2000. p. 59-62. [ Links ]
3. al-Mefty O, Marano G, Rajaraman S, Nugent GR, Rodean N. Transient ischemic attacks due to increased platelet aggregation and adhesivenness. Ultraestructural and functional correlation. J Neurosurg 1979;50:449-53. [ Links ]
4. Holliday PL, Mammen EF, Gilroy J, Buday J, Barnhart M. Sticky Platelet syndrome and cerebral Infarction in young adults. Ninth International Joint Conference on Stroke and Cerebral Circulation; 1983; Phoenix, AZ. Abstracts. [ Links ]
5. Rubenfire M, Blevins RD, Barnhart M, Housholder S, Selik N, Mammen EF. Platelet hyperaggregability in patients with chest pain and angiographically normal coronary arteries. Am J Cardiol 1986;57:657-60. [ Links ]
6. Mammen EF. Ten years experience with the sticky platelet syndrome. Clin Appl Thromb Hemost 1995;1:66-72. [ Links ]
7. Bick RL. Sticky platelet syndrome: a common cause of unexplained venous and arterial thrombosis. Results of prevalence and treatment outcome. Clin Appl Thromb Hemost 1998;4:77-81. [ Links ]
8. Mammen EF. Sticky platelet syndrome. Semin Thromb Hemost 1999;25:361-5. [ Links ]
9. Tofler GH, Brezinski D, Shafer AI, Czeisler CA, Rutherford JD, William SN, et al. Concurrent morning increase in platelet agreggability and the risk of miocardial infarction and sudden cardiac death. N Engl J Med 1987; 316:1514-8. [ Links ]
10. Chaturvedi S, Dzieczkowski JS. Protein S deficiency, activated protein C resistance and sticky platelet syndrome in a young woman with bilateral strokes. Cerebrovasc Dis 1999;9:127-30. [ Links ]
11. Sinzinger H, Kaliman J, O'Grady J. Platelet lypoxigenase defect (Wien Penzing defect) in two patients with myocardial infarction. Am J Hematol 1991;36:202-5. [ Links ]
12. Mammen EF, Comp PC, Gosselin R, Greenberg C, Hoots WK, Kessler C, et al. PFA-100 System: a new method for assessment of platelet dysfunction. Semin Thromb Hemostat 1998;24:195-202. [ Links ]
13. Bick R, Kaplan H. Syndromes of thrombosis and hypercoagulability: congenital and acquired thrombophilias. Clin Appl Thromb Hemost 1998;4:25-50. [ Links ]
14. Patrono C. Aspirin as an antiplatelet drug. N Engl J Med 1994;330:1287-94. [ Links ]
15. Beltrán E, Linares P, Castaño S. El síndrome de la plaqueta pegajosa. Primer reporte de casos en niños en Colombia y revisión de la literatura. Pediatría 2002;37:15-21. [ Links ]

