











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
Autoimmune pancreatitis (AIP) is a form of chronic pancreatitis whose main clinical manifestation is painless obstructive jaundice.1,2 It is a rare condition, with a prevalence of approximately 1 case per 100 000 people in the general population, accounting for 5-6% of all chronic pancreatitis cases.3,4 Based on its clinical manifestations and histologic pattern, AIP can be classified into two subtypes, namely, type 1 or lymphoplasmacytic sclerosing pancreatitis and type 2 or idiopathic ducto-centric pancreatitis4,5
In order to diagnose AIP, different aspects should be considered, among them: imaging findings -through computed axial tomography (CT) or magnetic resonance imaging (MRI)- of an enlarged diffuse pancreatic enlargement and loss of lobularity; serological findings such as elevated immunoglobulins; and histological findings such as lymphoplasmacytic infiltration without granulocytic acinar inflammation, storiform fibrosis, obliterative phlebitis, and presence of immunoglobulin G4 (IgG4), as well as involvement of other organs and positive response to corticosteroids.6-8 AIP does not require surgery and has a good prognosis with medical treatment.6
The following is the case of a patient with AIP diagnosed due to the presence of IgG4 and successfully treated with corticosteroid therapy. This case report was written following the CARE guideline criteria.9
Case presentation
A 62-year-old man, with no relevant medical history, visited the emergency room of a quaternary care hospital in Bogotá, Colombia, after 5 days of clinical symptoms consisting of generalized jaundice accompanied by choluria and episodes of acholia. Physical examination on admission revealed scleral icterus and generalized jaundice.
Upon admission, laboratory tests were performed showing blood count within normal ranges, preserved renal function (Cr: 0.8 mg/dL; BUN: 10 mg/dL), glucose at 108 mg/dL, and amylase levels within normal values (70 U/L). However, the liver profile was altered, with elevated levels of alkaline phosphatase (606 U/L), gamma-glutamyl transferase (1096 U/L), aspartate aminotransferase (AST) (223 U/L), alanine aminotransferase (ALT) (635 U/L), total bilirubin (13.5 mg/dL), direct bilirubin (10.6 mg/dL), and indirect bilirubin (2.9 mg/dL). Besides these tests, an abdominal ultrasound was performed, which showed intrahepatic and extrahe-patic biliary tract dilatation with radicals that reached 6mm, a common bile duct diameter of 10mm, as well as a hypoechoic focal lesion in the head of the pancreas with lobulated contours of approximately 33x30x42mm that did not show flow on color Doppler examination.
In view of these findings, the patient was admitted to the hospital and during his first day of stay he was assessed by the general surgery department, which considered that he presented hyperbilirubinemia of obstructive pattern with a lesion in the head of the pancreas. So, CT and MRI scans of the abdomen were requested, as well as tests for tumor markers. The test results were available on the following day and reported negative tumor markers (CA 19-9: 14.5 U/mL, alpha-fetoprotein: 2.46 ng/mL, and carcinoembryonic antigen: 2.19 ng/mL). The MRI showed a diffuse alteration in the morphology of the pancreas, with enlargement and obstruction of both the bile duct and the pancreatic duct (without dilatation) (Figure 1). Finally, the CT scan showed an alteration of the peripancreatic fat that formed a low-density "halo" sign and suggested AIP as the main differential diagnosis (Figure 2). Considering these findings, during the third day of hospital stay, the general surgery service considered ruling out an inflammatory process, and in view of a possible autoimmune etiology, requested immunoglobulin blood tests.
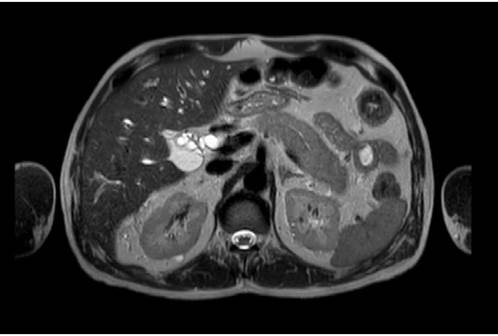
Source: Image obtained while conducting the study.
Figure 1 Magnetic resonance imaging of the abdomen showing alteration of the pancreatic morphology with enlargement and loss of lobularity.
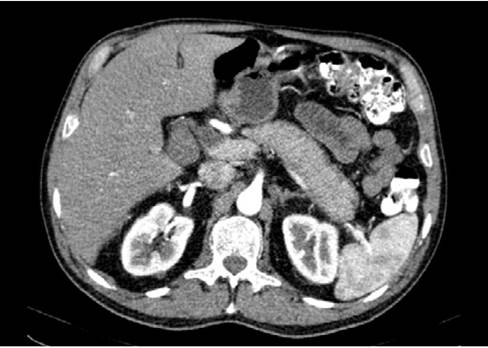
Source: Image obtained while conducting the study.
Figure 2 Computed axial tomography of the abdomen showing a low- density halo sign around the pancreas due to altered peripancreatic fat.
Since his admission, the patient was on intravenous fluid therapy, and on the fourth day of his hospital stay, he was evaluated by the gastroenterology department, which found a "pencil tip" amputation of the bile and pancreatic ducts above the papilla on the diagnostic scans. Due to the suspicion of bile duct stricture, an endoscopic retrograde cholangiopancreatography (ERCP) was performed the following day, confirming an intrapancreatic common bile duct stricture with a length of 1cm. On the basis of these findings, a 7x7cm plastic stent was placed during the procedure, successfully and completely closing the area of stricture. On the sixth day of hospital stay, the gastroenterology department requested an endosonography to rule out malignancy, and the report showed an enlarged pancreas with lobularity of the parenchyma without focal masses, thus corroborating the suspicion of AIP, and a biopsy was taken for histological study.
Accordingly, on the seventh day of hospitalization, the general surgery and gastroenterology services jointly decided to initiate corticosteroid therapy with prednisolone 40mg per day, which resulted in a significant improvement of the clinical condition of the patient three days after its instauration. Thus, 10 days after admission, given the adequate clinical course of the patient, it was decided to discharge him from the hospital, maintaining the outpatient treatment with prednisolone for 6 weeks at the aforementioned dose.
The patient attended the hospital 5 days after his discharge for a follow-up appointment with the general surgery service with evident improvement of his clinical condition and with the results of the immunoglobulin test, which showed elevated immunoglobulin levels (IgG1: 877 mg/dL, IgG2: 397 mg/dL, IgG3: 135 mg/dL, IgG4: 518 mg/dL), and the pathology report of the pancreatic biopsy that showed fragments of pancreatic parenchyma with loss of acinar cells, presence of ducts with reactive changes, and marked fibrosis with chronic lymphoplasmacytic inflammatory infiltrate, which are characteristic findings of chronic pancreatitis. Moreover, immunohistochemistry was positive for IgG, with 8 IgG4-positive cells with an IgG4/IgG ratio <10%, confirming the diagnosis of AIP. The patient continued on corticosteroid therapy at a dose of 40mg per day for 4 more weeks until completing 6 weeks of treatment. After 2 months, he attended a follow-up appointment in which it was found that his symptoms had resolved, and the disease was adequately controlled.
Discussion
AIP is a form of chronic pancreatitis characterized by clinical manifestations such as obstructive jaundice and histological changes of the pancreas (lymphoplasmacytic infiltration and fibrosis).1,2 Unlike other forms of chronic pancreatitis, this condition usually responds to corticosteroid therapy and is completely reversible in most cases.1,2 It was first described in 1961 when Sarles et al.10 reported a form of idiopathic chronic pancreatitis that was associated with jaundice and hypergammaglobulinemia, raising suspicion of an autoimmune process underlying this manifestation.1,2 However, it was not until 1995 when Yoshida et al.11 coined the term "autoimmune pancreatitis", which is still used worldwide to refer to this form of chronic pancreatitis3,4
AIP is considered to be a rare disease, with a prevalence of approximately 1 case per 100 000 people in the general population,12 accounting for 5-6% of all chronic pancreatitis cases.3,4 In clinical terms, AIP is characterized by painless obstructive jaundice, but its clinical spectrum varies widely, including manifestations of acute pancreatitis, onset of diabetes, weight loss, asthenia, and adynamia.4,5 To make a diagnosis, in 2010, the International Association of Pancreatology developed the International consensus diagnostic criteria (ICDC) for AIP,13 which established that aspects such as imaging, serologic and histologic findings, involvement of other organs, and response to corticosteroids should all be considered together.5,7
Regarding imaging findings, it has been reported that CT and MRI scans in these patients show a diffuse enlargement of the pancreas (sausage-shaped appearance) associated with a loss of loburality, as well as a hypoattenuated peripheral halo sign on CT and hypointensity on MRI. During ERCP, it is common to see a long stricture (>1/3 of the pancreatic duct) and multiple or focal strictures without proximal dilatation of the system.5,7 Serologic findings involve elevated levels of immunoglobulins, especially IgG4, antinuclear antibodies, and rheumatoid factor; however, it should be noted that, in some cases of AIP, immunoglobulin levels are not elevated.8
In their study, Cai & Tan7 state that AIP may cause extrapancreatic lesions due to the involvement of other organs, including lacrimal and salivary gland lesions, pulmonary lesions (involving hilar lymphadenopathy), sclerosing cholangitis, tubulointerstitial nephritis, and retroperitoneal fibrosis. Likewise, it has been established that histological findings include lymphoplasmacytic infiltration with or without granulocytes, storiform fibrosis, obliterative phlebitis, and presence of IgG4.6-8
Regarding the criterion of response to corticosteroids, O'Reilly et al.1 state that the initiation of corticosteroid therapy has shown resolution of both imaging and clinical manifestations in patients with AIP. Consequently, response to treatment is also considered as a diagnostic criterion.
In the present case, the patient was diagnosed with AIP since both CT and MRI scans showed a diffuse alteration of the pancreatic morphology, an enlargement of the pancreas and an obstruction of the bile duct and pancreatic duct (suggesting narrowing of these organs), with no findings of proximal dilatation, which are characteristic imaging findings of this condition.1,2
With respect to serological criteria, the literature cites multiple alterations, including elevated IgG levels,4,5 which is key to diagnose AIP and to exclude other differential diagnoses. It has also been established that IgG4 levels >135 mg/dL can differentiate AIP from pancreatic cancer, with a limited sensitivity ranging from 79% to 93%. In addition, IgG4 levels >280 mg/dL are considered to have a specificity of 99% for AIP,1,2 so the serological criterion was met in the present case since the patient's IgG4 level was 518 mg/dL.
Regarding the criterion of involvement of other organs, it has been concluded that the most affected organs are the biliary tree, the lymph nodes, and the salivary and lacrimal glands.1,4 In the present case, this parameter was not observed; however, it is considered that its occurrence cannot be ruled out because, according to the literature, extrapancreatic manifestations can be prior, simultaneous or even subsequent to AIP.2,4,5
In order to describe the histological parameter, it is necessary to point out that, according to the presentation of the diagnostic criteria for AIP established by the ICDC, it can be classified into two subtypes: type 1 AIP and type 2 AIP. Type 1, or lymphoplasmacytic sclerosing pancreatitis due to its histologic pattern, occurs more frequently in men, with a peak incidence during the sixth decade of life, and is currently recognized as the pancreatic manifestation of IgG4-related disease because it causes abundant plasma cell infiltration (IgG4 >10 cells/field), stromal fibrosis, obliterative phlebitis with pancreatic inflammation, and elevated serum IgG4 levels, and it also has an excellent response to corticosteroid therapy.5-7 On the other hand, type 2, or idiopathic duct-centric pancreatitis, does not cause elevated serum IgG4 levels and its histopathologic features include granulocytic epithelial lesions with little or no plasma cell infiltration (IgG4 <10 cells/field).2,4 In the present case, the biopsy result is striking as it reported 8 IgG4-positive cells, a characteristic finding of type 2 AIP, contrary to type 1 AIP where IgG4-positive cells are usually >10.
Finally, given that, as mentioned above, corticosteroids are not only used to treat this disease, but a positive response to them is also a diagnostic criterion of AIP,6,7 it is recommended to administer prednisone or prednisolone at 0.6 mg/kg per day for 4 to 6 weeks.6 In the present case, given the high suspicion of AIP, prednisolone was administered at a dose of 40mg per day for 6 weeks, which resulted in an improvement, so it is considered that the patient met the criteria of responding to corticosteroid therapy. Thus, in association with the criteria of the symptoms mentioned above, it can be concluded that the patient presented type 1 AIP, since the observed findings and his clinical course are characteristic of this subtype, despite the fact that the biopsy showed only 8 IgG4-positive cells.
Conclusions
AIP is a disease that every general practitioner and specialist should be aware of and consider as a differential diagnosis in patients with symptoms of painless obstructive jaundice and imaging findings of a diffusely enlarged pancreas and loss of lobularity, thus avoiding misdiagnosis and unnecessary interventions.

