











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
Sitosterolemia is an autosomal recessive disease caused by mutations in the ABCG5 or ABCG8 genes, encoding the sterol-1 and sterol-2 proteins, respectively, which work as obligate heterodimers to pump sterols out of hepatocytes, vesicular epithelium and enterocytes, and are responsible for intestinal and biliary excretion of cholesterol and phytosterols.1-3 Therefore, a disorder of these genes triggers decreased hepatic and intestinal excretion of plant sterols, resulting in their accumulation in blood and tissues2,4,5and leading to the development of hypercholes -terolemia and xanthomas. 2,6,7
Sitosterolemia is considered a rare disease, with only 110 cases reported worldwide by Myrie et al.2 in 2020. Likewise, the frequency of mutations changes depending on the population: while mutations in the ABCG5 gene are associated with Asian patients, mutations in the ABCG8 gene are associated with Caucasian patients. 6
Sitosterolemia was first described in 1974 under the name ß-sitosterolemia by Bhattacharyya & Connor, 8 who reported the case of two Caucasian sisters (both college students) who had elevated plasma levels of phytosterols accompanied by tendon xanthomas since childhood.
This article reports the case of a patient with non-consanguineous parents, who was diagnosed with sitosterolemia by means of next-generation gene sequencing (NGS) analysis of the ABCG5 and ABCG8 genes. It should be noted that, to date, this is the first case of this disease reported in Colombia.
Case presentation
5-year-old male, born and raised in Bogotá (Colombia), son of non-consanguineous parents. In April 2021, he was taken for the first time to the Human Genetics outpatient service of a healthcare institution in Bogotá in order to establish his diagnosis since he presented with xanthomatosis with no established etiology, had elevated levels of low-density lipoprotein (LDL cholesterol), and a diagnosis of familial hypercholesterolemia had been ruled out as a possible cause of xanthomas.
According to his parents, at the age of 4 years the patient began to develop bilateral xanthomas in knees, elbows and Achilles tendon, and his total and LDL cholesterol levels were outside normal limits (up to 970mg/dL and 810mg/dL, respectively) despite pharmacological treatment with ezetimibe 10mg every day and cholestyramine 4g every 8 hours, both orally. No significant family history was reported, as neither his sister (daughter of the same parents) nor his parents had dyslipidemia.
Physical examination revealed the following findings: weight of 14kg (-2.65 z score), height of 97.5cm (-3.06 z score), head circumference of 55cm (1.4 z score), normal vital signs, and non-dysmorphic phenotype. In addition, xanthomas were observed on knees, elbows, ankles, and heels (Figure 1). No other abnormal findings were reported.
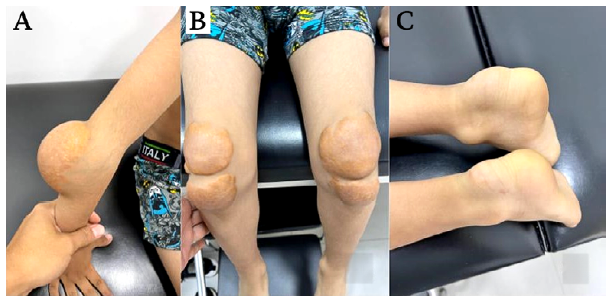
Source: Images obtained while conducting the study.
Figure 1 Xanthomas in elbows (A), knees (b) and Achilles tendon (C).
The referring service (pediatric endocrinology) ordered laboratory tests, which were performed on April 20, 2021. The patient's mother took the results to the first consultation with the human genetics service in order to initiate follow-up by that service (Table 1). Due to the extremely high levels of total and LDL cholesterol reported in the baseline tests, it was decided to continue with pharmacological treatment without modification with ezetimibe and cholestyramine.
Table 1 Biochemistry laboratory tests of the patient.

LDL: low-density lipoproteins; HDL: high-density lipoproteins; AST: aspartate aminotransferase; ALT: alanine aminotransferase; TSH: thyroid-stimulating hormone; Free T4: free thyroxine; N/A: not applicable.
Source: Own elaboration.
Considering the patient's age, the presence of multiple large xanthomas, the extremely high levels of total and LDL cholesterol, and that familial hypercholesterolemia had been ruled out, sitosterolemia was suspected. Therefore, in the first consultation with the human genetics service, NGS of the genes involved in sterol metabolism was requested. The results were available at the follow-up appointment with this service in June 2021 and are presented in Table 2.
Table 2 Next-generation genomic sequencing plus analysis of deletions and duplications for genes ABCA1, ABCG5, ABCG8, ANGPTL3, APOA1, APOA5, APOB, APOC2, CETP, CREB3L3, CYP27A1, GPD1, GPIHBP1, LCAT, LDLR, LDLRAP1, LIPA, LIPG, LMF1, LPL, LRP6, MTTP, PCSK9, PNPLA2, SAR1B, APOA4, APOC3, CYP7A1, GALNT2, GCKR, LIPC, LIPI, MYLIP, PLTP, SCARB1, and ZHX3.
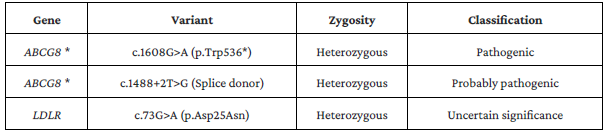
* Data on the variants in ABCG8 do not allow a definitive determination of whether these variants are on the same chromosome or on opposite chromosomes. LDL receptor (LDLR).
Source: Own elaboration.
NGS results indicated that the c.1608G>A (p.Trp536*) and c.1488+2T>G (Splice donor) variants of the ABCG8 gene are pathogenic and probably pathogenic, respectively. This demonstrated an alteration in the conformation of the ABCG8 protein that affected sterol excretion and confirmed the diagnosis of sitosterolemia.
Once the diagnosis was confirmed, recommendations were given to the patient and his family during the June 2021 check-up. First, the parents were suggested to put the child on a diet low in plant sterols (oils, nuts, avocado, and chocolate) and seafood sterols, and the patient was referred to the nutrition service to design a diet to suit his condition. Secondly, continuation of the previously established pharmacological treatment was indicated. Finally, it was suggested to continue performing periodic liver function tests, considering that this disease may affect the liver and the hypertransaminasemia observed in the latest tests.
In the last follow-up visit with the human genetics service (March 2022), it was evident that the transaminase values improved compared with those obtained 8 months earlier (June 2021), being much lower (Table 2). Likewise, total cholesterol, LDL and triglyceride values had a notable decrease, with no adverse effects reported by the patient or his parents.
Despite the improvement in the laboratory tests, no significant changes were observed in the xanthomas, so it was decided to continue with the monitoring by the pediatric endocrinology service, as well as the dietary recommendations and pharmacological treatment to control the levels of sterols in the blood and achieve an improvement or resolution of these lesions, which can be treated by surgical excision if they do not show a reduction in size. The measures implemented were effective and in a follow-up with pediatric endocrinology, 9 months after the last follow-up with human genetics (December 2022), there was evidence of a decrease in xanthomas, so a surgical intervention was ruled out.
At the time of writing this case report, the patient had a satisfactory progress and, in order to provide adequate follow-up, he continued to be treated by the pediatric endocrinology group of the institution and following the recommendations provided in the June 2021 follow-up, which initially led to endocrinological improvement and later to an improvement of the xanthomas.
Discussion
Sitosterolemia is an autosomal recessively inherited disease caused by variations in the ABCG5 or ABCG8 genes, leading to inactivation of sterolin-1 and sterolin-2, which decreases sterol excretion.4-6 The approximate prevalence of this metabolic disorder in the general population is more than 1 case per 200 000 people, 4,6,9 being higher in residents of Kosrae (Micronesia, United States; up to 13%), in Hutterites and Amish people (up to 8% and 4%, respectively), and in populations of Chinese, Japanese and Indian origin.2
Sitosterolemia has a wide phenotypic heterogeneity. 7 The manifestations of the patient of the case described here were severe hypercholesterolemia, which is mostly observed in children, 4 and xanthomas in unusual locations, such as elbows, heels and knees, which had already been described by Nuñez-Rodriguez et al.5 Other frequent manifestations of sitosterolemia, but absent in the reported patient, include: arthralgia, progressive liver disease, premature atherosclerosis, 2,4 and hematologic disorders such as thrombocytopenia with macrothrombocytopenia due to elevated thrombopoietin levels. 2-6,10
The diagnostic criteria for sitosterolemia (Table 3) take into account clinical manifestations, laboratory test results, exclusion of differential diagnoses, and genetic analysis of the ABCG5 and ABCG8 genes. 9

In the case reported, at the first visit, the presence of xanthomas and familial hypercholesterolemia previously ruled out by the absence of pathogenic variants of the LDLR, APOB and PCSK911 genes confirmed the diagnostic criteria A-1 and C, respectively. Therefore, NGS was requested to establish whether the genetic criterion (D) was present, as it determines the diagnosis of sitosterolemia. In this patient, phytosterol levels were not measured because false negatives have been reported in subjects with diets low in phytosterols and on treatment with ezetimibe or bile acid sequestrants, 2 rendering criterion B invalid due to the patient's treatment with ezetimibe and cholestyramine.
When phenotypic and laboratory findings suggest a diagnosis of sitosterolemia, a multigene panel should be used as a genetic method of diagnostic confirmation. 2 Another method is exome sequencing, although its implementation is discouraged since the diagnosis of this disease has not been established using this method in any case report. 2
In the present case, NGS was chosen, detecting the c.1608G>A (p.Trp536*) variant that creates a premature translation stop codon (p.Trp536*) in the ABCG8 gene and, consequently, resulting in a protein product that is absent or altered in its functions. NGS also detected the c.1488+2T>G (splice donor) variant affecting a donor splice site in intron 10 of the ABCG8 gene, which disrupts RNA binding and results in loss of protein function. These findings of mutations in the ABCG8 gene confirmed the diagnosis of sitosterolemia (criterion D).
It should be noted that the presence of simultaneous pathogenic variants in the ABCG5 and ABCG8 genes is rare and has only been reported once. 7
Conventional treatment of sitosterolemia consists of dietary changes and pharmacological therapy. 2 Regarding diet, the main measure is the restriction of phytosterol consumption (corn, sesame and rice oil, peanuts, margarine, avocado, and chocolate) and cholesterol-rich foods, 4,9 which was implemented in our patient. In turn, pharmacological treatment for sitosterolemia, which was also implemented in this case, involves the combined use of bile acid sequestrants (cholestyramine) and sterol absorption inhibitors (ezetimibe) to reduce sitosterol and LDL levels. 2,9,12
Statins, in principle, are not a therapeutic option because the enzyme hydroxy-meth-yl-glutaryl-CoA reductase (HMG-CoA reductase) is down-regulated. 12,13 However, in cases of sitosterolemia with advanced atherosclerosis, statins can be added to the treatment, and if they are not effective, LDL apheresis is indicated. 9 Another option reported in the literature is liver transplantation, which was performed in the case of a patient with sitosterolemia and cirrhosis, producing a dramatic reduction in the levels of phytosterols. However, that patient did not require additional interventions to the conventional ones (dietary changes and use of cholestyramine plus ezetimibe), such as statins or liver transplantation, since there was no evidence of advanced atherosclerosis and there was an improvement in the levels of transaminases. 14
Xanthomas usually resolve with conventional treatment, 2 as in the present case, in which the patient improved in that aspect after more than one year of treatment. If xanthomas do not improve or produce disability, surgical excision may be considered. 15
Finally, it should be noted that, unlike familial hypercholesterolemia, patients with sitosterolemia respond favorably to a reduction in the consumption of phytosterols, 9 even though they are indicated for a healthy population16,17 since they reduce their cardiovascular risk. Also, in the present case, there was evidence of improvement in the lipid profile and resolution of the xanthomas after the synergy of pharmacological treatment and dietary changes.
Conclusions
Sitosterolemia is a rare inherited disease that should be considered in pediatric patients with multiple xanthomas and in whom familial hypercholesterolemia has been ruled out. Therefore, performing NGS of the genes involved is important to make a timely diagnosis and initiate appropriate treatment, which will improve the prognosis and quality of life of these patients.
The present case, the first of its kind reported in Colombia, provides information on the clinical characteristics of sitosterolemia in the country, as well as the diagnostic approach that should be considered in patients with multiple xanthomatosis or premature coronary artery disease in order to reduce underdiagnosis rates. This will allow early diagnosis, initiate timely treatment and, consequently, avoid adverse outcomes.

