











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Basal ganglia calcifications may be a radiological finding in approximately 20% of the general population, and a variety of medical conditions have been documented in which they may be present, both idiopathically as well as secondary to other disorders 1,2. This is the case of Fahr's disease, which is a rare neurodegenerative disorder on which very little has been published in Colombia 3. It consists of bilateral idiopathic basal ganglia calcifications that are expressed clinically with neuropsychiatric manifestations and/or motor symptoms. Normally, the age of onset is between 30 and 60 years, with gradual symptom progression. No correlation has been found between the age of onset, the extent of calcium deposits and the neurological deficits 4. Over the years, it has also been known as idiopathic basal ganglia calcification, bilateral strio-pallido-dentate calcinosis (BSPDC), and primary familial brain calification (PFBC) 2. On the other hand, the name "Fahr's syndrome" is used when the disease has an identifiable and potentially treatable etiology; it has been associated mainly with endocrine diseases affecting calcium and phosphorus metabolism, like hypoparathyroidism, but also to infections, neuroinfections, exposure to toxins, and immune diseases, among others 2.
We present the case of a patient who had choreiform movements as the only clinical manifestation and a tomographic finding of extensive basal ganglia calcifications.
Case presentation
This case involved a 69-year-old female mixed-race patient from Tubará, Atlántico, who lived in the township of Galapa, Atlántico. She was admitted due to the sudden onset of stereotyped, broad, arrhythmic, purposeless movements of all four extremities and the orolingual region, predominantly on the right side of the body, which did not recede during sleep. She had a history of type 2 diabetes mellitus being treated with recently instated basal-bolus insulin at a total daily dose of 36 units, metformin 850 mg/day and glibenclamide 5 mg/every 12 hours.
On physical exam, she was in good general condition, her vital signs were within normal limits, she was alert and oriented with pupils equal and reactive to light, and she had preserved muscle strength in all four extremities, normal reflexes and no sensory deficit. Motor evaluation showed involuntary, arrhythmic, erratic, broad movements of the upper and lower limbs, predominantly on the right side, which did not affect her grip and were described as chorea.
Initial laboratory tests were ordered showing the following: complete blood count (CBC), serum electrolytes, kidney function, coagulation times, bilirubin, cholesterol and transaminases within normal limits; blood glucose in the hypoglycemic range, and elevated glycosylated hemoglobin. The results were as follows: CBC: leukocytes 4,740/uL, red blood cells 4,620,000/uL, hemoglobin 12.7 g/dL, hematocrit 35.6%, mean corpuscular volume 76.9 fL, mean corpuscular hemoglobin 27.4 pg, mean corpuscular hemoglobin concentration 35.5 g/dL, red cell distribution width 13.1%, red cell distribution width 2.84 g/dL, platelets 211 x 103 /uL, mean platelet volume 8.0 fL, platelet distribution width 59.4%, percentage of granulocytes 67.0%, percentage of lymphocytes 24.7%, percentage of monocytes 4.4%, percentage of eosinophils 1.5%, percentage of basophils 0.4%, percentage of large unstained cells (LUCs) 2.0, total granulocyte count 3.18 x103 /uL, total lymphocyte count 1.17 x103 /uL, monocyte count 0.21 x103 /uL, eosinophil count 0.07 x103 / uL, basophil count 0.02 x103 /uL, LUC count 0.09 x103 /uL; serum potassium: 4.16 meq/L; serum sodium: 139.6 meq/L; blood urea nitrogen: 11.21 mg/dL; urea: 24 mg/dL; serum creatinine: 1.28 mg/dL; microalbuminuria: 10 mg/L; partial thromboplastin time: 25.7 sec; prothrombin time: 10.3 sec; total bilirubin: 0.96 mg/dL; direct bilirubin: 0.43 mg/dL; indirect bilirubin: 0.53 mg/dL; aspartate aminotransaminase (AST): 15 U/L; alanine aminotransaminase (ALT): 21 U/L; total cholesterol: 208 mg/dL; HDL cholesterol: 49.5 mg/dL; LDL cholesterol: 128 mg/dL; triglycerides: 145 mg/dL; glycosylated hemoglobin: 6.4%; and blood glucose: 38 mg/dL.
A simple head tomography showed signs of mild cortical atrophy. There were bilateral ring-shaped hyperdensities with irregular borders in the brain stem, basal ganglia and lateral ventricles, involving the cerebellar cortex, especially of the left hemisphere, the head of the caudate nucleus, the internal capsule and the lentiform nucleus, predominantly in the right hemisphere (Figure 1). Symptom management was started with oral haloperidol at 2.5 mg every eight hours and the oral hypoglycemics were deferred, achieving blood glucose control. Calcium and thyroid homeostasis tests were ordered to rule out an organic cause, finding normal vitamin D, ionized calcium, phosphorus, magnesium, thyroid stimulating hormone (TSH) and parathyroid hormone (PTH) levels; free thyroxine (fT4): 1.14 ng/dL; TSH: 1.772 IU/mL; ionized calcium: 1.17 mmol/L; 25-hydroxy vitamin D: 47.6 ng/mL; phosphorus: 3.71 mg/dL; magnesium: 2.03 mg/dL; and PTH: 37.7 pg/mL. Due to persistent symptoms during her hospital stay, the initial dose was adjusted to 5 mg in the morning and 2.5 mg at night, with an adequate clinical response.
Considering the lateralization of the symptoms and the sudden onset of the condition, magnetic resonance imaging was ordered to rule out an ischemic cause, with the following findings: changes in signal intensity in the basal ganglia and both cerebellar lobes, related to mineral and/or calcium deposit disease; chronic microangiopathic changes; moderate cortical atrophy; and peripheral thickening of the left maxillary sinus mucosa. These results were corroborated with the previously mentioned simple head CT results, thus determining a diagnosis of Fahr's disease. After ruling out secondary causes of the signs and symptoms described, and considering the results of imaging studies, she was discharged on haloperidol at the dose which provided a clinical response, her glucose-lowering treatment was adjusted, and she was given a follow up appointment in one month in the outpatient department, at which she showed improvement of her initial condition and adequate metabolic control (Figure 2).
Discussion
According to the existing literature, Fahr's disease manifests clinically as heterogenous neurological disorders (clumsiness, fatigue, unstable gait, slow or dragging speech, involuntary movements, bradykinesia, rigidity, propulsive gate, low voice volume, mask-like facies, dystonia, tremors, choreoathetosis, seizures, and pyramidal or cerebellar signs), psychiatric disorders (cognitive impairment, mood disorders, psychotic symptoms and obsessive-compulsive symptoms) and, radiologically, as bilateral calcifications, predominantly of the basal ganglia 2,3. This agrees with this patient's clinical picture, as she had choreiform movements of all four extremities which were much wider on the right side of her body. She did not have psychiatric symptoms; however, given the neuroprogressive nature of the condition, their future appearance cannot be ruled out.
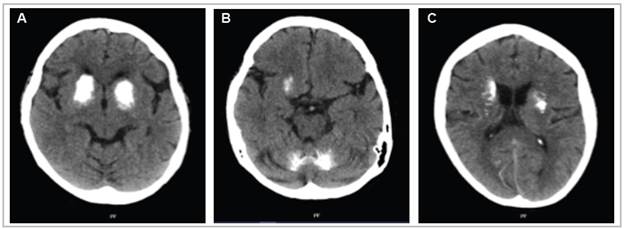
Figure 1 A simple axial head tomography showing marked intraparenchymal calcifications occupying almost all the basal ganglia, bilaterally, and the central region of the cerebellar lobes.

Figure 2 Magnetic resonance imaging of the brain in the T2 (A), Diffusion (B), and T2 GRE (C,D) sequences of the axial plane. Hypointensity is seen bilaterally in the head of the caudate and putamen due to ferromagnetic material deposits (A), hyperintensity in the same locations (B), symmetrical hyperintensity in the basal ganglia (C), and hyperintensity in the dentate nuclei of the cerebellum (D).
To arrive at this diagnosis, the current evidence of the disease was used, consisting of bilateral basal ganglia calcifications, progressive neurological deterioration, and the absence of metabolic disorders, other infectious diseases, trauma, toxins or hereditary/familial disorders. In this case, the first radiological resource used was a head CT without contrast, which is the tool with the greatest sensitivity and specificity for detecting calcifications 5,6,8.
In patients with psychotic syndrome, it is very important to rule out an organic etiology, as there are case reports 7 indicating that some patients debut with isolated acute-onset psychiatric symptoms. Therefore, when there is a slight suspicion of an organic etiology or when the patient's symptoms are refractory to various levels of treatment, neuroimaging must be used to provide the correct clinical approach for the patient and a more favorable prognosis.
There is no currently available treatment to delay or reverse the course of the disease, and therefore its prognosis varies. Treatment is only symptomatic: anticonvulsants, antipsychotics, mood stabilizers like lithium carbonate, clonazepam, and antidepressants have proven effective in several case reports 1,3, especially in patients with predominantly neuropsychiatric symptoms. In our case, haloperidol was able to decrease the frequency and amplitude of the choreiform movements. Given the patient's clinical improvement and adequate response to medication, she was discharged on the same dose of haloperidol.
Some questions remain in this case, one of which is whether the presentation was due to a sporadic appearance of the disease or was linked to autosomal dominant or recessive inheritance. Mutations of the SCL20A2, PDGFB, PDGFRB and XPR1 genes have been reported to be involved, and therefore molecular genetics studies are needed to determine the etiology. In addition, it is vitally important to provide genetic counseling for close relatives as, if there is a high risk of the disease, screening head CTs have been proposed 5.
Much remains to be studied in this disease, especially the development of effective medications to prevent its neuroprogression and all the catastrophic consequences for the quality of life of those who suffer from it. It remains for us, as healthcare professionals, to suspect and correctly identify this condition, and provide symptomatic treatment to the patients, along with proper multidisciplinary counseling to enable them to better carry out their activities of daily living.

