











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
El sector apícola está relacionado con el desarrollo natural de los ecosistemas y con el avance económico a nivel agrícola y pecuario, puesto que tiene función protagónica en la polinización y, por tanto, en la sustentabilidad de la producción agrícola. Del mismo modo, es importante por el valor nutricional de sus productos. Dada su importancia para la agricultura, su desarrollo es imprescindible para alcanzar la seguridad y soberanía alimentaria que requiere el país (producción de alimentos, cantidad, calidad, inocuidad, trazabilidad y sostenibilidad), pues a medida que se incrementa el desarrollo apícola, se aporta mayor beneficio ambiental, económico y social para todo el sector agrícola. Pese a su importancia, la apicultura enfrenta importantes retos productivos causados, principalmente, por el efecto antrópico en los ecosistemas y el cambio climático 1.
Siguiendo con la importancia de la apicultura, gracias a la polinización entomófila se mantiene aproximadamente un tercio de los cultivos y la mayor parte de la flora silvestre 2. Se calcula que en los últimos años la polinización ha representado un beneficio económico global alrededor de los 265 mil millones de euros, correspondientes al precio de las cosechas que dependen de la polinización natural; se estima que sin esta herramienta se bajaría la productividad agrícola hasta en un 75% 2.
Así bien, para efectuar el proceso de polinización, entre 10000 y 25000 abejas obreras realizan en promedio diez viajes por día, para lo cual exploran aproximadamente un radio de 3 km en los alrededores de su hábitat 3,4. Durante este proceso, microorganismos, sustancias químicas y partículas en el aire entran en contacto con ellas 5. Por lo anterior, las abejas y los productos de la colmena sirven como bioindicadores para el monitoreo de plagas, contaminación de origen industrial o urbano y/o prácticas agrícolas que puedan ser perjudiciales 6.
Entre dichas prácticas agrícolas se encuentra el control químico de plagas, de mayor impacto para el ecosistema, pues trae como principal consecuencia un aumento en la frecuencia y dosis de exposición a pesticidas por parte de las abejas. Por lo anterior, la actual regulación para plaguicidas químicos no solo está orientada a la protección del consumidor, sino que también busca la protección de las abejas como principales polinizadores en la naturaleza 2.
Ahora bien, el análisis de residuos contaminantes orgánicos en productos apícolas se realiza mediante dos tipos de métodos: los presuntivos y los de confirmación. En el primer grupo se encuentran los inmunoensayos y los métodos microbiológicos. En el segundo los métodos de confirmación que, según el Codex Alimentarius, se basan en técnicas instrumentales avanzadas como la electroforesis capilar y la cromatografía líquida de alta eficiencia acopladas a espectrometría de masas (HPLC-MS) 7,8 Diferentes entes de control como la Administración de medicamentos y alimentos (FDA, por sus siglas en inglés) y la Unión Europea han establecido la necesidad de contar con métodos que permitan realizar la cuantificación y confirmación de los residuos 7 , 8,21,22, por lo cual los métodos instrumentales tienen mayor ventaja en este caso 9-11.
En el análisis de muestras de miel, debido a la complejidad de la matriz y a que es necesario realizar exhaustivos procesos de limpieza, se han reportado diversos métodos, entre ellos el QuEChERS modificado 12. Aunque este método resulta ser una excelente alternativa cuando se dispone de analizadores de masas en tándem, al emplear detectores convencionales como fluorescencia, nitrógeno-fósforo, captura de electrones o inclusive un espectrometrómetro de masas con un cuadrupolo sencillo, la cantidad de interferentes limita el uso del método en un número elevado de compuestos o impide la obtención de límites de cuantificación acordes con la legislación 13,14.
En este sentido, este trabajo tuvo como objetivo desarrollar un método para la determinación de 56 plaguicidas de diferentes familias químicas en muestras de miel, mediante el empleo de UFLC (cromatógrafo líquido ultra rápido por sus siglas eninglés) acoplado a un analizador de masas de cuadrupolo simple. De igual modo, se buscó demostrar la aptitud del método mediante una validación de nivel 2 15.
Materiales y métodos
Materiales, reactivos y soluciones
Se trabajó con estándares de plaguicidas de pureza mayor al 95%, provenientes de las casas comerciales Dr Ehrenstorfer y Chemservice. Las soluciones madre fueron preparadas en concentraciones cercanas a 500 μg/mL en acetonitrilo o metanol y almacenadas en frascos ámbar a -20 °C. Los solventes empleados en este estudio fueron J.T. Baker grado HPLC. Para los ensayos de extracción se utilizaron sales de QuEChERS Restek Q-Sep TM y para la limpieza de los extractos se emplearon los adsorbentes Restek dSPE Q-Sep TM. Los detalles de preparación mezclas y del estándar interno se encuentran en trabajos previos 16,17.
Mieles de partida
Se emplearon cuatro mieles obtenidas de apiarios experimentales y mieles comerciales provenientes del altiplano cundiboyacense y del departamento del Meta en Colombia. Todo el material fue analizado antes y durante todo el proceso de desarrollo del método analítico, con el propósito de asegurar que no se tuviera presencia de ninguno de los plaguicidas estudiados, así como de controlar los procesos de contaminación cruzada.
Método de extracción de partida
Se partió del método QuEChERS para mieles 18. En un tubo de centrífuga se pesaron 5 g de muestra, se adicionó TPP (fosfato de trifenilo) y la mezcla de plaguicidas (concentraciones entre 0,1 y 5 μg/mL). Se dejó en reposo por 15 min y, transcurrido ese tiempo, se adicionaron 10 mL de agua y 10 mL de acetonitrilo; se agitó de manera manual por 1 min y, posteriormente, se adicionaron 4 g de MgSO4 anhidro y 1 g de AcONa; nuevamente se agitó manualmente por 1 min. Después, se centrifugó a 6000 rpm por 5 min y se tomaron 10 mL del sobrenadante, que se transfirió a un tubo de centrífuga de 15 mL
Para el proceso de limpieza, por cada mililitro de extracto se adicionaron 25 mg de amina primaria/secundaria (PSA, por sus siglas en inglés) y 150 mg de MgSO4 anhidro, se agitó manualmente por 30 s y se centrifugó por 2 min a 6000 rpm. Se tomaron 5 mL de extracto y se concentraron con un flujo de nitrogéno hasta llevar a casi sequedad, posteriormente se adicionaron 20 μL del El y se reconstituyó con 1 mL de una mezcla Agua-ACN (7:3). Finalmente, el sobrenadante se filtró a través de una membrana de 0,22 μm de PTFE y se transfirió a un vial de cromatografía 19. Durante el desarrollo del método, en todos los experimentos se realizó un mínimo de cinco réplicas.
Estudio de los adsorbentes
Una vez establecidas las mejores condiciones en la etapa de extracción de los compuestos, con el propósito de mejorar la selectividad del método, se procedió a estudiar el empleo de diferentes adsorbentes para la limpieza de los extractos. De acuerdo a la composición química de las mieles, se considera que los principales compuestos que interfieren en el proceso de detección de los plaguicidas pueden ser vitaminas, fenoles, pigmentos, aminoácidos, ácidos orgánicos e inclusive azúcares 28. La reducción de la presencia de ácidos, aminoácidos y azúcares, como se mencionó en la sección experimental, se realizó a través del empleo de PSA y sulfato de magnesio. De esta manera, para mejorar el proceso de limpieza se adicionaron otros adsorbentes: octadecilsilano (C18; 20 mg por cada mL de extracto) y carbón negro grafitizado (CNG; 20 mg por cada mL).
Calibración analítica y análisis de datos
Se realizó un proceso de evaluación de efecto matriz previo a la cuantificación de cada analito. Se encontraron porcentajes de efecto matriz entre 69% y 130%, resultados que concuerdan con estudios previos 17 , 20. Por lo anterior se realizaron también diferentes niveles de calibración en blancos de matriz. Para el caso de los ensayos de recuperación, se cuantificó con un solo punto de calibración, de acuerdo a las recomendaciones del documento SANTE 20. Para la evaluación de la linealidad del sistema, se preparon las curvas de calibración que estaban compuestas por seis puntos de calibración en blanco de matriz, cada uno de ellos se inyectó cuatro veces en el sistema analítico; los puntos se distribuyeron equidistantemente. Todos los análisis estadísticos se realizaron a través del paquete estadístico R Commander a un nivel de confianza del 95% 22.
Validación del sistema de medición.
Con el fin de demostrar que el sistema de medición funcionaba adecuadamente, se realizó una validación de nivel 2, consistente en evaluar: selectividad, exactitud, linealidad, límites de detección y límites de cuantificación.
Por otro lado, para la evaluación de la exactitud del método, como recuperación y precisión en condiciones de repetibilidad y de precisión intermedia, se fortificaron muestras de miel a tres niveles de concentración. El primer nivel (LC) correspondía al límite máximo de residuos, una concentración inferior a este o en su defecto concentraciones cercanas a 10 μg/kg. El segundo y tercer nivel de fortificación correspondieron a 2 y 5 veces el primer nivel (LC).
Condiciones cromatográficas
El análisis cromatográfico fue llevado a cabo en un UFLC Shimadzu Prominence (Maryland, CA, EUA), acoplado a un detector selectivo de masas LCMS-2020. Los análisis fueron llevados a cabo en una columna Shim Pack (6 cm x 2 mm d.i., tamaño de partícula de 2,1 μm y fase estacionaria C18); se trabajó en modo de gradiente con ácido fórmico al 0,1% (p/v) y acetato de amonio 5 mM en agua Milli-Q (A); la fase orgánica empleada fue acetonitrilo (B). El programa de elución utilizado, expresado como porcentaje de B, inicia a 0% (0 min) aumenta a 20% en 0,01 min, se mantiene por 0,3 min, posteriormente llega hasta 55% en 8,5 min y luego aumenta hasta el 100% en los siguientes 1,5 min. Finalmente, se mantiene por 1,5 min 17. El volumen de inyección fue 5 μL, la temperatura de columna 40 °C y el flujo de fase móvil 0,3 mL/min.
Condiciones de la interfaz y espectrómetro de masas
Con el propósito de obtener una mejor sensibilidad para la identificación y cuantificación de los 56 plaguicidas en estudio, se trabajó con dos métodos de adquisición independientes: un estándar interno (TBP) y un estándar subrogado (TPP) Aunque las condiciones cromatográficas fueron las mismas, las condiciones de la interfaz y del espectrómetro variaron. En este sentido, el equipo operó en modo electrospray, con un flujo de gas de secado de 10 L/ min y un flujo de gas nebulizador de 1,5 L/min; las temperaturas del bloque de calentamiento y de la línea de eliminación del solvente correspondieron a 200 °C y 250 °C y a 210 °C y 250 °C para el primer (Grupo 1 de compuestos) y segundo método de adquisición (Grupo 2 de compuestos), respectivamente 16,17.
Los análisis fueron llevados de manera simultánea en modo positivo y negativo, el voltaje aplicado en el capilar correspondió a 4500 V y -4500 V respectivamente. Todos los análisis fueron realizados en modo SIM. La Tabla 1 muestra los iones seleccionados para la cuantificación y el tiempo de retención de cada compuesto: las tres primeras columnas muestran las condiciones para el método de adquisición 1, mientras que las tres últimas columnas son para el método de adquisición 2. Estos compuestos fueron seleccionados acorde con la legislación nacional e internacional 23, el nivel de afectación a las abejas, su uso en Colombia, sensibilidad en el instrumento y la disponibilidad del material de referencia para la calibración del instrumento.

Resultados y discusión
Estudio de la relación cantidad de muestra/solvente de extracción
Con el propósito de obtener límites de cuantificación acordes con la legislación existente para mieles 23, se partió del método europeo (citratos) y se realizaron ensayos en los que se incrementó la cantidad de muestra desde 3 g hasta 10 g y otros en los que se disminuyó la cantidad de solvente de 10 mL a 5 mL.
Se encontró que los porcentajes de recuperación de varios compuestos como el metalaxil, acefato, oxamilo, entre otros, disminuyeron notablemente. Asimismo, la cantidad de interferencias que se obtuvieron al incrementar la relación muestra/solvente de extracción impidió la detección adecuada de varios compuestos. En este sentido, como parte del desarrollo del método y en vista de los resultados obtenidos en esta primera etapa se decidió trabajar con 5 g de muestra y 10 mL de solvente orgánico e incluir una etapa de concentración de la muestra.
Selección del sistema de amortiguación de pH
Debido a que el método QuEChERS presenta dos versiones, reconocidas como métodos oficiales para el análisis de residuos de plaguicidas, se evaluaron las dos con el propósito de determinar cuál es la más adecuada. La primera corresponde al método AOAC 2007.01 19 (Método A), en la que se usa acetato de sodio para realizar el control de pH en el proceso de extracción. La segunda versión del método considera el uso de sales de citrato y corresponde a la versión europea del método EN 15662 26, (método B).
Los resultados mostraron que, mediante el método A, 14 compuestos presentaron interferencias que impidieron realizar la integración de las señales cromatográficas; para el método B se presentaron 13 interferencias. Lo anterior supone que, debido a una baja selectividad de los métodos, no es posible la cuantificación de cerca del 24% de los compuestos bajo estudio. La Figura 1 muestra los compuestos que presentaron interferencias y una recuperación inferior al 70% en al menos uno de los métodos. Por otro lado, los compuestos restantes presentaron recuperaciones entre el 70% y el 120%, recuperación que se considera aceptable para este tipo de determinaciones 21.
Se realizó un análisis de varianza con el propósito de evaluar si existen diferencias significativas (α = 0,05) entre las medias de los porcentajes de recuperación. Los resultados no fueron del todo concluyentes, pues para la mayoría de los casos no se presentaron diferencias significativas entre los sistemas evaluados. Sin embargo, al comparar con los criterios de la Unión Europea, se encuentra que sí se observan diferencias, por ejemplo, para dinotefuran se encontró una probabilidad de aceptar H0 mayor a 0,05. No hay diferencias significativas, pero en la Figura 1 se encuentra que, con el método B, este compuesto cumple con lo establecido por la Unión Europea. Por este motivo, los análisis estadísticos se omitieron en este estudio y todos los resultados y discusión se centran en lo establecido en los criterios de la Unión Europea para este tipo de métodos analíticos.

Figura 1 Comparación de los porcentajes de recuperación al emplear acetato o citrato como sal de control de pH
Como se observa en la Figura 1, existen casos críticos en los que la recuperación se encuentra por debajo del 50%, por ejemplo espinosad (A y D) y carbendazim; los demás compuestos presentan valores de recuperación muy cercanos al 70 %. Por otro lado, en la evaluación de las precisiones de cada uno de los métodos, se encontró que mediante el método A, se presentaron coeficientes de variación superiores al 15% en 11 casos, mientras que para el método B se encontraron 8 casos.
El método B mostró una mejor exactitud, reflejada en mejor selectividad, mayor número de compuestos con recuperación aceptable y menor dispersión en los resultados.
Los resultados indicaron que, para el caso de los plaguicidas estudiados y la matriz de interés, resulta más conveniente realizar la extracción con las sales de citrato, es decir empleando el método B. Sin embargo, es de resaltar que la versión europea del método, además de las sales de citrato, incluye NaCl, lo cual incrementa la fuerza iónica y, por consiguiente, mejora la transferencia de los analitos a la fase orgánica. Por lo anterior, se decidió trabajar con este sistema amortiguador de pH.
Adición de las sales
El método original indica que las sales de citrato, el NaCl y el MgSO4, se adicionan una vez se agrega el agua a la miel y se homogeniza esta mezcla. Durante este proceso, se observó que las sales se aglomeraban y su dispersión a través de la mezcla de extracción (miel-agua-acetonitrilo) no era sencilla de realizar. Adicionalmente, hubo un calentamiento significativo, debido a la hidratación del MgSO4, por lo cual se decidió disolver las sales de citrato y NaCl para realizar su adición junto con el agua. Entonces, en lugar de adicionar sólo agua para el proceso de homogenización de la sal, se adicionaron 10 mL de una solución amortiguadora de citratos (pH = 6,8), la cual tenía la cantidad de sales equivalente al método QuEChERS, a excepción del MgSO 4 .
Posterior a esta adición, se realizó una homogenización de la mezcla, se adicionó el acetonitrilo y se realizó la extracción con agitación por 10 min en un vórtex, luego se destapó el tubo y se realizó la adición del MgSO 4 ; se agitó manualmente. Algunos de los resultados obtenidos a través de estas modificaciones se presentan en la Figura 2. Se encontró que las modificaciones realizadas al método mejoran los resultados en la exactitud del método de manera notable.
La Figura 2 muestra que, para 8 de los 12 compuestos que no se recuperaron con el método tradicional, se obtuvieron porcentajes superiores al 70%, lo cual indica que el sesgo del método disminuyó. Sin embargo, de acuerdo a estos resultados, carbendazim, tiodicarb, dazomet y espinosad (A y D) siguen presentando porcentajes de recuperación bajos.
La mejora en la recuperación se atribuye a varios factores: por un lado, el proceso de extracción se realizó sin la presencia del MgSO4 en la mezcla de la miel, puesto que este se adiciona hasta el final de este proceso; adicionalmente, al realizar la extracción con solo la disolución amortiguadora y el acetonitrilo, se obtiene un sistema más homogéneo (no hay aglomeraciones) y no se dan procesos de calentamiento por la adición del MgSO4 en el momento de la extracción. Por otro lado, posiblemente se tenga un mejor control del pH al adicionar la disolución amortiguadora, de lo contrario, solo hasta que se disolviera la totalidad de las sales de citrato en la mezcla, se lograría el control del pH debido a la viscosidad de la miel.
Sumado a lo anterior, cuando se realiza la adición del MgSO4 a la mezcla (al final de la extracción), el calor liberado parece disiparse más rápidamente pues, si bien se forma la aglomeración del método original, esta es más fácil y rápida de disolver a través de la agitación manual. Se cree que el uso del vórtex también contribuye al aumento de los porcentajes de recuperación pues facilita la transferencia de masa del analito hacia la fase orgánica y se aplica durante un mayor tiempo, lo que garantiza que exista un equilibrio mayor. Finalmente, las modificaciones realizadas al método no redujeron la cantidad de compuestos con interferencias, es decir, con este método se obtuvo el mismo número de compuestos que no se pudieron determinar debido a problemas de selectividad. Incluso, en los perfiles cromatográficos obtenidos para los blancos con el uso de este método, se observó una mayor cantidad de ruido en comparación con el método original. Lo anterior es atribuido a las modificaciones realizadas, pues junto con los analitos existe una mayor cantidad de compuestos de la matriz que se coextraen.

pH de disolución amortiguadora
En diferentes trabajos se encuentran estudios relacionados con la influencia del pH sobre la recuperación de algunos compuestos 26,27. Sin embargo, en todos los estudios se menciona el término pH aparente, pues esta magnitud es medida en acetonitrilo y no en una solución acuosa. En este sentido y acorde con la instrumentación y materiales de referencia con los que se cuenta para la calibración de los instrumentos de pH, se optó por evaluar la influencia del pH de la disolución de amortiguación sobre la exactitud de los analitos. Para ello se ajustaron las disoluciones con ácido fórmico a 4 diferentes valores de pH; la Figura 3 muestra un resumen de los resultados obtenidos. Allí se presenta el promedio de recuperación de todos los compuestos, el número de compuestos que tienen porcentajes de recuperación inferiores a 70% y el número de compuestos para los que el método no es preciso, es decir, porcentajes de coeficiente de variación superiores al 15%.

Figura 3 Porcentajes de recuperación promedio (%R), número de compuestos con %R inferiores a 70% y con coeficientes de variación (%CV) mayores al 15% para los diferentes valores de pH
La Figura 3 muestra que al comparar los tres valores más altos de pH, se encuentra que estos no varían de manera considerable, igual que el número de compuestos con recuperaciones bajas o dispersiones altas. Sin embargo, para los dos valores de pH más bajos se observa que, tanto la recuperación como la precisión del método, es incrementada, pues tienen solo 6 y 2 compuestos con % CV mayores al 15%; para el valor de pH más bajo se tienen solo 3 compuestos con recuperaciones inferiores al 70%.
El incremento de la precisión y de la recuperación de los compuestos a pH bajos, se atribuye a una mayor estabilidad de los compuestos a estos valores de pH 25. En diferentes reportes se encuentra que el valor de "pH aparente" (en acetonitrilo), donde la extracción de los plaguicidas fue más eficiente, se encuentra entre 5,5 y 5,8. Esto indica que, posiblemente, el resultado de la mezcla de la miel con las disoluciones amortiguadoras de valores de pH más bajos (5 y 5,5) proporcionan valores de pH en el sistema de extracción (miel + disolución) similar a este valor de pH aparente, reportado en otros estudios 19.
La estabilidad de los compuestos no solo puede originar errores sistemáticos (como menores porcentajes de recuperación), sino también puede originar errores aleatorios: pequeños cambios en otras magnitudes de influencia como la temperatura o el tiempo de extracción suponen un aumento de estos errores. En este sentido, el mejor promedio en los porcentajes de recuperación de los compuestos se logra con el pH más bajo, valor al que la mayoría de los compuestos presentan los menores errores sistemáticos.
Sin embargo, en este valor de pH, cualquier mínimo cambio en otra variable de influencia genera un aumento en los errores aleatorios, por lo cual se presentan mayores coeficientes de variación que para el valor de pH de 5,5. De esta manera, al realizar la extracción a un pH de 5,5 se tiene un punto de equilibrio entre el control del error sistemático y el error aleatorio, pues, como se puede observar en la Figura 3, es donde se tiene una mejor exactitud. Por lo tanto, a partir de este ensayo se concluye que, dentro de los valores de pH evaluados, la mejor condición corresponde a 5,5.
Finalmente, los compuestos que presentaron porcentajes de recuperación menores al 70% a pH 5 correspondieron a los isómeros del espinosad y carbendazim. Para el caso de los primeros no fue posible encontrar los valores de pKa, por lo cual se cree que estos compuestos sufren algún tipo de hidrólisis o descomposición que impide una adecuada extracción. Para el caso del carbendazim, el pKa es 4,48, por tanto, es posible que al encontrarse la molécula ionizada la extracción por parte de un solvente orgánico no sea eficiente 19.
Adsorbentes de limpieza
La Tabla 2 muestra un resumen de los resultados obtenidos a través del uso de diferentes sistemas de limpieza. Es de aclarar que para este estudio se partió de blancos de miel, se obtuvieron extractos libres de plaguicidas, se mezclaron y fueron fortificados antes del proceso de limpieza, lo cual buscaba reducir la variación producida por el proceso de extracción 29.
Tabla 2 Porcentajes de recuperación obtenidos en la evaluación de diferentes sistemas de limpieza

I: Compuesto con interferencia que no permitió determinare! porcentaje de recuperación
La Tabla 2 evidencia que el empleo de otro tipo de adsorbentes mejora la selectividad de los compuestos, pues se observa un descenso en el número de compuestos con interferentes, respecto a la limpieza únicamente con PSA para cualquiera de los sistemas evaluados. También se observa que la mejor alternativa para la limpieza de los extractos es el C18, pues mejoró la selectividad para casi la totalidad de los compuestos en los dos conjuntos de ensayos en los que se usó.
Para el caso de tiociclam y de metil azinfos, no se pudo mejorar la selectividad del método, a pesar de que se realizaron algunos experimentos adicionales en los que se aumentó la cantidad de adsorbentes usados; en su lugar se obtuvieron porcentajes de recuperación menores para algunos compuestos.
Para el caso específico del Tiociclam, debido a su naturaleza polar (ver tiempos de retención en la Tabla 1), se cree que es necesario realizar otro tipo de limpieza como cromatografía de intercambio iónico, de columna en fase normal o la adición de otro adsorbente más polar como la sílica, pues el interferente debe ser de la misma naturaleza. Para el caso del metil azinfos se considera que la interferencia de origen tiene una estructura muy similar a este compuesto.
Por otra parte, se encuentra que el uso de CNG no contribuye de manera significativa a la mejora de la selectividad del método, pues los resultados indican que permite la reducción de interferentes para sólo tres compuestos. Asimismo, al observar el último sistema que incluye C18 y PSA, se encuentra que el carbón no aporta de manera significativa, al contrario, para algunos compuestos los porcentajes de recuperación son inferiores, lo cual se debe posiblemente a la adsorción de los analitos generada por la mayor área superficial que se tiene.
El acetonitrilo es un solvente conocido por no extraer demasiados compuestos de naturaleza lipídica así como compuestos de elevada polaridad como el caso de los azúcares.
Sin embargo, el aumento de la fuerza iónica en el sistema de extracción a través del MgSO 4 y del NaCl, inducen a que se extraigan este tipo de compuestos junto con los analitos. En este sentido, se considera que el empleo de C18 reduce notablemente la cantidad de interferentes, debido a la adsorción de este tipo de compuestos que tienen baja solubilidad en acetonitrilo. Adicionalmente, es posible queexista un efecto sinérgico al hacer uso de PSA y C18, pues las señales producidas por los interferentes químicos se redujeron a lo largo de todo el cromatograma. Finalmente, la Figura 4 describe el esquema general del método desarrollado.

Validación del sistema de medición
La selectividad del método se evaluó a través del uso de cuatro diferentes tipos de miel, los cuales se diferenciaban por su origen. Los resultados obtenidos en esta evaluación mostraron resultados idénticos a los reportados en la Tabla 2 (sistema PSA + C18), es decir, el método es selectivo para todos los analitos de interés excepto para metil azinfos y tiociclam.
En las Tablas 3 y 4 se presentan las concentraciones de fortificación, los resultados de exactitud y los límites de detección del método. Los límites de detección reportados, fueron estimados a partir de diferentes aproximaciones (30) y para todos los casos en el proceso de confirmación se encontró que la relación señal/ruido era superior a 3.
Tabla 3 Resultados obtenidos en la validación del método desarrollado. Grupo de compuestos 1- Método de adquisición 1
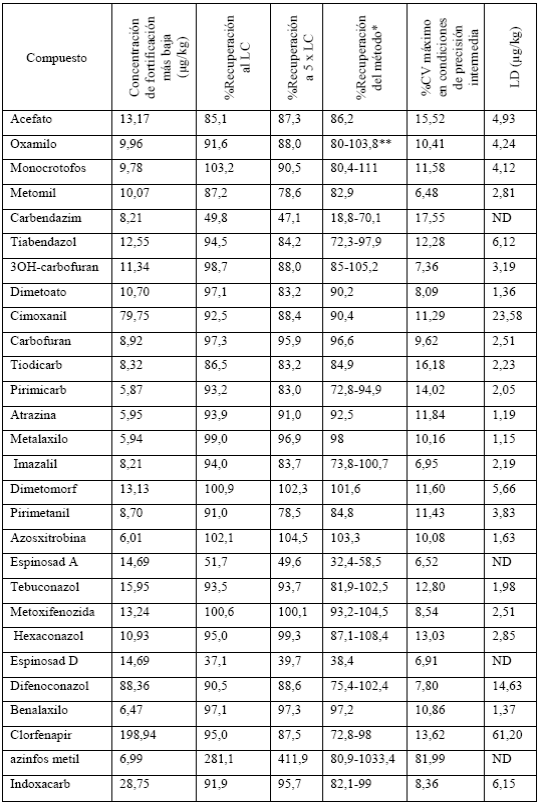
*Estimado a partir de los resultados de precisión intermedia. Para los casos en los que se presentó heterocedasticidad entre las diferentes concentraciones evaluadas se presenta el intervalo de porcentajes de recuperación. ND: No determinado para este compuesto.
Tabla 4 Resultados obtenidos en la validación del método desarrollado. Grupo de compuestos 2- Método de adquisición 2

*Estimado a partir de los resultados de precisión intermedia. Para los casos en los que se presentó heterocedasticidad entre las diferentes concentraciones evaluadas se presenta el intervalo de porcentajes de recuperación. ND: No determinado para este compuesto.
Las Tablas 3 y 4 muestran que para la mayoría de los compuestos se presentaron porcentajes de recuperación entre el 70% y 120%, por lo cual se puede concluir que el método presenta un error sistemático aceptable, a excepción de carbendazim, espinosad (A y D), lufenuron y piraclostrobina, cuyos porcentajes de recuperación fueron inferiores al 70%.
Por otro lado, se encontró que el máximo %CV para cada uno de los analitos, en general, es inferior al 15%, lo cual cumple con el criterio de la Unión Europea 21. Sin embargo, para algunos compuestos se observa que este %CV supera el 15%, pero debe tenerse en cuenta que el criterio de la Unión Europea se establece para compuestos en condiciones de repetibilidad, y que el %CV presentado en las Tablas 3 y 4 se obtiene para condiciones de precisión intermedia (interanalistas). Por lo tanto, se considera que para todos los casos el método es preciso, a excepción de los casos en los que el método no es selectivo (tiociclam y metil azinfos) o presenta una recuperación inadecuada (menor al 70%).
La evaluación de la linealidad del sistema de medición se realizó a través de diferentes pruebas como: ANOVA, falta de ajuste, significancia de la pendiente y del intercepto, entre otras 16. Todos los resultados indicaron que el sistema es lineal en el intervalo de concentraciones presentadas en la Tabla 5.

Conclusiones
Se logró desarrollar y validar un método para el análisis de residuos de plaguicidas en mieles, basado en una extracción líquido-líquido con acetonitrilo y posterior limpieza a través de extracción en fase sólida dispersiva. En el proceso de desarrollo del método, se encontró que la homogenización de la miel con una solución amortiguadora a un pH de 5,5 incrementa la exactitud del método.
Por otro lado, la selectividad del método mejoró notablemente al realizar uso de una mezcla de octadecilsilano (C18) y sílica funcionalizada con aminas (PSA). Por su parte, el proceso de validación indicó que el método es selectivo, preciso, lineal y exacto para 50 de los 56 plaguicidas empleados. Finalmente, se encontró que el método no es selectivo para 2 de los compuestos estudiados y no es exacto para 4.

