











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
The synthesis, characterization, and identification of molecular systems potentially useful in the design of molecular machines is still a huge challenge for supramolecular chemistry researchers 1 , 2. In this regard, some organic compounds with double bonds (C=C, C=N, or N=N) are of special interest due to their reversible photochemical and thermal reactions. Such feature makes these compounds potentially useful for the synthesis of molecular machines 3 , 4. It is well known that hydrazone derivatives are one of the most used compounds as building blocks for the formation of supramolecular systems because of the physical and chemical properties that they exhibit. These properties can be reversibly modulated in response to an external stimulus, either light 5 - 9, addition of metal ions 10 - 14, and pH changes 15. Hydrazone derivatives based on 2-quinoline are particularly interesting molecules for the development of photo-activated switches as, upon UV-light irradiation, the quinoline-nitrogen may form an intramolecular hydrogen bond with the N-H proton from a hydrazine moiety, thus stabilizing the Z isomer making even possible to isolate it and study its properties. Additionally, the E-isomer can act as a ligand similar to terpydine for metal-ion coordination 16 , 17.
Likewise, the study of electronic and electrochemical properties of configurational isomers has not been well documented, at least for hydrazone derivatives, where there are only a few recent reports 7 , 18. Electrochemistry of E/Z configurational isomers is important to demonstrate their potential use as molecular photoswitches and electronic devices. Therefore, to design molecular systems able to reversibly modulate the physical and chemical properties in response to light or metal centers, we have decided to study and compare through theoretical studies the electronic and electrochemical properties of the configurational isomers of 2-((2-(4-chlorophenylhydrazono)methyl)-quinoline.
Materials and Methods
All starting reagents were acquired from Sigma-Aldrich (USA) and were used without additional purification. FT-IR, NMR (mono and bidimensional), and UV-vis spectra were taken in a Shimadzu FTIR-8400S instrument (Japan), in a 400 MHz Bruker Ultrashield spectrometer (USA), and in a PharmaSpec Shimadzu UV-Vis UV-1700 spectrophotometer (Japan), respectively. Electrochemical measurements were recorded in a bipotentiostat model 700B (USA) series electrochemical Analyzer/ Workstation from CH Instruments coupled to a computer.
Synthesis of hydrazone derivatives
The hydrazone derivative (E)-2-((2-(4-Chlorophenylhydrazono)methyl) quinoline 1-E was synthesized following the same synthetic protocol as reported in the literature 7. A mixture of 2-methylquinoline (200 mg, 1.40 mmol) and selenium dioxide (263.5 mg, 2.37 mmol) was dissolved in dry dioxane (20 mL). Then, the reaction mixture was heated under reflux and monitored by thin-layer chromatography (TLC). After disappearance of the starting material, a solid was obtained which was filtered and washed with chloroform (3 x 5 mL). The 2-quinolinecarboxaldehyde was purified by column chromatography with a 95:5 CHCl3/MeOH solution getting an 80% yield.
The aldehyde derivative was condensed with 4-chloropehnylhydrazine which was added in a 1:1 ratio in absolute ethanol and heated for 3 h until a red precipitate was formed. This precipitate was washed with cold ethanol and recrystallized from ethanol in 82% yield. Mp 206-208 °C. 205-207191H NMR (400 MHz, DMSO-d6) δ 7.34-7.41 (m, 4H), 7.75 (t, 1 H, J 7.61), 7.91-8.02 (m, 1 H), 8.16 (d, 1 H, J 8.00), 8.25 (d, 1 H, J 8.39), 8.338.44 (m, 2 H), 8.72 (d, 1 H, J 8.98), 12.43 (s, 1 H); 13C NMR (100 MHz, DMSO-d6) δ 115.8. 118.4, 122.6, 125.9, 126.6, 127.4, 128.4, 128.8, 129.1, 129.7, 133.2, 142.03, 142.8, 152.5.
(Z)-2-((2-(4-chlorophenylhydrazono)methyl)quinoline (1-Z)
A solution of 1-E in methanol was irradiated with a 250 Watts mercury lamp for 30 min. This compound was purified by column chromatography using CHCl3 as eluent obtaining a yellow oil; yield 65%; 1H NMR (400 MHz, DMSO-d6) δ 7.22-7.35 (m, 4H), 7.58-7.64 (m, 1H), 7.80 (t, 1H, J 7.7 Hz), 7.99-8.05 (m, 2H), 8.11-8.21 (m, 2H), 8.44 (d, 1H, J 8.98 Hz), 11.62 (s, 1H).
Single-Crystal structure determination
The data were collected on a Bruker APEX-II CCD diffractometer using MoKα radiation (0.71073 Å) monochromated by graphite at room temperature (296 K). The cell determination and the final cell parameters were acquired on all reflections using the Bruker SAINT software 20, included in APEX2 software suite. Data integration and scaled was carried out using the Bruker SAINT software 20 .
The structure was solved using the SHELXS-2013 software and refined using SHELXL-2013 (21;, contained in WinGX 22, and Olex2-1.2 23. Non-hydrogen atoms of the molecules were clearly resolved and full-matrix least-squares refinements of these atoms with anisotropic thermal parameters were performed. All hydrogen atoms were stereochemically positioned and refined with the riding model 23, except for the hydrogen atom bonded to Nitrogen atom, which was found from the density map. ORTEP diagram was generated with Olex223. Mercury software 24 was used to prepare the illustrations.
Electrochemical study of hydrazone derivatives
Cyclic voltammetry (CV) and Osteryoung Square Wave Voltammetry (OSWV) were performed using a 0.1 M solution of tetrabutylammonium hexafluorophosphate (TBAPF6) in DMF. A glassy carbon electrode, a platinum wire, and a silver wire were used as working, counter, and pseudo-reference electrode, respectively. The solutions were degassed with argon before each measurement (10 min). Ferrocene was added as an internal standard. The scan rate was 100 mVs-1.
Computational details
Theoretical calculations were performed using GaussView as graphic interface 24 and Gaussian 09 26 for running calculations. Molecular geometry of 1-E in the ground state was optimized by DFT:B3LYP method with 6-311+G (d, p) basis set. NMR chemical shifts were computed at the same level of theory, using the gauge-including atomic orbital (GIAO) method without any solvent or solvation effect considerations.
Vibrational frequencies were performed at the same level than vibrational analysis and assuring that global minima were achieved. Hartree-Fock/6-31+G (d) level of theory and Self-Consistent Isodensity Polarized Continuum Model (SCI-PCM) were employed to simulate solvation effect in optimizations, while UV-Vis spectrum calculations were carried out using same method and level of theory without solvation. For UV-Vis spectrum deconvolution, theoretical IR, and UV spectra graph generation GaussSum 3.0 software package was employed 27.
Results and Discussion
Synthesis and characterization of hydrazone derivatives
Details of the synthesis of 1-E and 1-Z are shown in Figure 1. Compound 1-E was synthesized by the partial oxidation of 2 with selenium dioxide in dioxane, followed by condensation of 3 with the hydrazine derivative 4 in ethanol 7. The condensation reaction led to the formation of the corresponding E-hydrazone, which was identified by the singlet at 12.43 ppm corresponding to the NH proton 3. The geometry optimization for 1 was also performed to compare the energy associated to each isomer, as it was expected the E isomer is 12.2 kJ/mol more stable than the Z isomer. The irradiation of 1-E with a 250 Watts mercury lamp produced the Z isomer, which was isolated by column chromatography in a 67% yield.

For 1-Z, the 1H-NMR spectra show the existence of an intramolecular H-bond after irradiation with UV light since all the signals changed their chemical shift. The lack of symmetry in the molecule is reflected by the different coupling between the signals and the shift of the N-H proton in 1-Z when compared to 1-E (See Figure 2). The most remarkable change occurs on the NH and phenyl protons, which are shifted to the up-field region and splitted as a multiplet, respectively.

Single-crystal structure determination
Crystals of 1-E were obtained by slow evaporation of ethanol, resulting in fine, orange needlelike crystals. 1-E crystalized in the monoclinic space group P21/c. The molecule presents an almost planar configuration with a torsion angle between C6-C1-N1- N2 of 5.89(1)° and a dihedral angle between the quinoline and benzene planes of 1.91°. The Mogul geometry check shows that all bond lengths and angles are within the normal range 28. ORTEP representation of the 1-E structure is presented in the Figure 3a; Crystal data collection and structure refinement details are summarized in Table 1.
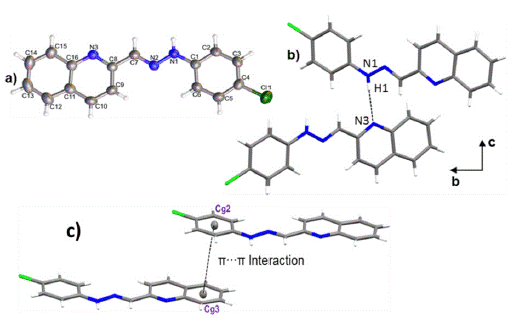
Figure 3 a) ORTEP representation of title compound, b) Hydrogen bond N1-H1-N3 and c) a) π·π interaction between Cg2 and Cg3.

The crystal packing is driven by N1-H1-N3 hydrogen bond, along [010] direction with 3.0855(1) Å of distance. As shown in Figure 3b, the interactions give rise to zig-zag chains growing along the [010] direction and the connected molecules are placed on the two planes (931) and (1241). A 3D supramolecular network is formed by the joining of the chains along [100] direction by π-π interactions between the aromatic rings with an intercentroids distance of Cg2-Cg3= 3.8903(1) Å (Figure 3 c). It was also observed a C9-H9---C11 interaction 29 along [001] direction with a distance of 3.7168(15) Å.
The Hirshfeld surface for 1-E showed that the chloride and the deprotonated nitrogen atoms in the quinoline and hydrazone present mainly, a hydrogen acceptor behavior, while the N-H and C-H act as donor groups (see Figure 4). Furthermore, an observation over the aromatic rings in the shape index surface blue and red regions that indicates the presence of π-π interactions, is confirmed by the fingerprint plot. The greatest contribution comes from Van der Waals interactions (H---H and C---H) followed by hydrogen bonds as C l ---H and N---H (see Figure 4b), that contribute with 15.6% and 7.9%, respectively. The π-π interactions contribute with 10.9% . Other contacts were also observed, however, their contribution is less than 5% each.

Molecular geometry optimization
Ab initio calculations yielded a planar structure with a dihedral angle equals to 0.001° between the quinoline and benzene planes and  torsion angles between the quinoline or benzene rings and the hydrazine group. As shown in Figure 5, these dihedral and torsional angles are not significantly deviated concerning to the results discussed in the crystallographic section.
torsion angles between the quinoline or benzene rings and the hydrazine group. As shown in Figure 5, these dihedral and torsional angles are not significantly deviated concerning to the results discussed in the crystallographic section.
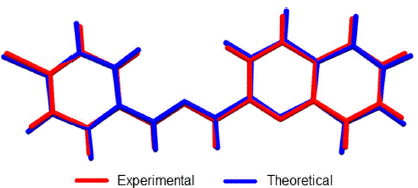
Figure 5 Overlap of crystallographic and optimized (DFT: B3LY-P/6-311+G (d, p) structures of 1-E. RMSD was 0.0985.
Linear regression analysis was also carried out to establish a systematic comparison between the optimized and crystallographic structures. The slope represents a ratio between the set of variations of calculated-experimental data. The intercept corresponds to the value of the dependent variable (calculated) when the independent variable (experimental) is equal to zero: meaning an over or underestimation value. Thus, perfect correlation is given when the slope and intercept values are 1 and 0, respectively (selected structural parameters and linear regression are shown in Table 2). The analysis reveals that theoretical calculations tend to a subtle overestimation respect to the crystallographic data set as shown in the intercept values (see Table 2).

The highest deviation from experimental bond length measurements was observed in N1-H bond (17.17% error) since this group is highly influenced by hydrogen bond interaction (See Figure 3). However, other bond angles and length show less than 1.4% and 0.9% bond length and angle errors respectively.
UV-Vis Spectroscopy analysis
The electronic transitions of 1-E were studied by UV-Vis. As shown in Figure 6, the hydrazone derivative exhibits a high absorption band around 425 nm, which present a bathochromic shift with the increasing polarity of the solvent. Additionally, a new band around 380 nm corresponding to π-π* transitions related to the imine bond was observed using methanol as solvent 6. An optimized structure by DFT: B3LYP/6-311+G (d, p) calculation was used as input geometry for a second optimization at HF/6-31+G (d) level of theory including solvent effects through the SCI-PCM approximation to simulate chloroform solvation yielding an equivalent conformation with no differences in the electronic structure. From this optimized structure, TD-SCF method was employed to calculate excited states energy at HF/6-31+G (d) level of theory neglecting solvation. Besides, the theoretical UV-Visible spectrum presents a transition in the visible region and two transitions in the ultraviolet region (absorptions at 427, 319, and 278 nm respectively) with an absolute error percentage respect to experimental observations equal to 6.57, 10.34, and 11.15%, respectively. These results tend to an underestimation in a range between 28 to 33 nm.

GaussSum 3.0 software package was employed to deconvolute the computed electronic transitions using Gaussian 09 and to determine the contribution of the main electronic transitions (see Figure 7). Higher oscillator strength was observed at 427 nm signal (0.3465). This absorption maximum was characterized by a main electronic transition from HOMO to LUMO+1 energy levels (34%), where a homogeneous electronic distribution is displaced to a MO over the hydrazone and phenyl groups.

A second main electronic transition from HOMO-2 to LUMO+1 (5%) was also observed. Second higher oscillator strength was observed at 278 nm (0.1705), where the main electronic transition (HOMO-1 ?LUMO+5, 21%) is characterized by a partial electronic displacement from the quino-line to hydrazone and chlorophenyl moieties.
NMR and FT-IR spectroscopy analysis
Theoretical-experimental comparison of FT-IR and NMR spectra of 1-E was also carried out. The four most prominent bands are C=N stretching (1579 cm-1), N-H rocking (1508 cm-1), N-N stretching (1142 cm-1), and C-H bending (1241cm-1). Displacement vectors for these vibrations are shown in Figure 8.
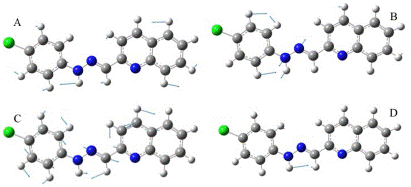
Figure 8 Displacement vectors for selected vibrational modes. A) C-H bending. B) N-N stretching. C) C=N stretching. D) N-H bending.
These vibrational modes are degenerated except N-H bending since displacement vectors clearly showed other vibrational modes coexisting simultaneously in each frequency.
The absence of imaginary frequencies confirms that the optimized structure is a global minimum. A comparison of selected computed vibrational frequencies and assigned values experimentally is shown in Table 3 from supporting information. Calculated values tend to overestimation even though the scale factor used is less than 1.00 30. The maximum error rate was observed in C-Cl frequency (5.45%), C=N and N-H frequencies are the second and third biggest deviation respect to experimental observations. C-Cl and N-H errors may be explained because these moieties are typically involved in hydrogen bonding interactions in the solid state and those were neglected in theoretical calculations.

The correlation between the computed and experimental 1H-NMR chemical shifts was unacceptable as the signal of N-H, showed the biggest deviation (see Table 4), mainly due to its high influence by solvation 31. Therefore, the neglected solvation effect should help to improve the quality of 1H-NMR chemical shift calculations. The 13C-NMR chemical shifts showed acceptable correlation respect to experimental values yielding an average rate of error equal to 2.61%.

Electrochemical studies
The electrochemical properties of compounds 1-E and 1-Z were studied by cyclic voltammetry (CV) and Osteryoung Square Wave Voltammetry (OSWV) (See Materials and Methods Section).
When analyzing the cathodic potentials (see Figure 9), three irreversible reduction potentials for the 1-E isomer are observed. These potentials remained irreversible upon increasing scan rates. OSWV revealed two additional reductions (see Figure 10) which are located very close to those observed in CV. Presumably, the closed peak potentials correspond to the processes occurring on the surface of the electrode and the corresponding cathodically shifted potential to the process occurring in the solution. The first reduction in compound 1-E is located at -1.09 V and corresponds to the imine reduction whereas for 1-Z is cathodically shifted towards -2.54 V (see Table 5). These results indicate that upon reduction the [1-E]' - radical anion is stabilized by π delocalization in the quinoline system. However, in the Z configuration the nitrogen from the quinoline is acting as a Lewis base (forming the intramolecular hydrogen bond) and, therefore, its participation in the radical anion stabilization is restricted thus shifting cathodically the reduction potential. Likewise, the differences in the current intensity for the cathodic/anodic potentials suggest different reduction mechanism for both configurational isomers.
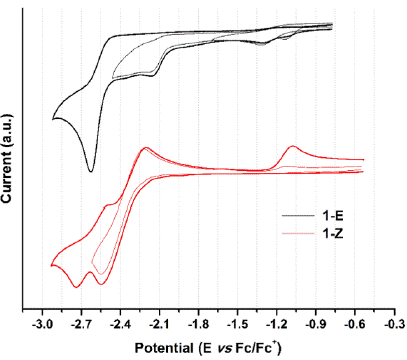
Figure 9 Cyclic voltammogram of hydrazone configurational isomers at 100 mVs-1. Glassy carbon as working electrode and ferrocene as an internal reference.
Table 5 Electrochemical ootentials of hydrazone derivatives vs ferrocene in DMF (V)a.
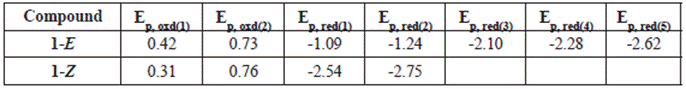
aWorking electrode: glassy carbon; counter electrode: Pt wire; Pseudoreference electrode: Ag wire. Supporting electrolyte: TBAPF6.Scan rate: 0.1 V s-1.
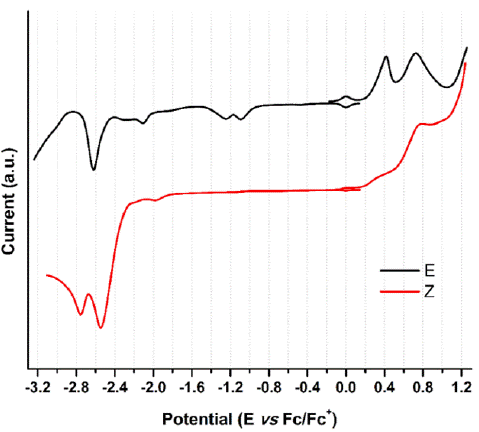
Figure 10 OSW of hydrazone derivatives 1-E and 1-Z at 100 mVs-1. Glassy carbon as working electrode and using ferrocene as an internal reference.
Compounds 1-E and 1-Z exhibited two irreversible oxidation potentials which are attributed to the -NH groups of the hydrazone framework. The first reduction potential of 1-E is anodically shifted, when compared to its 1-Z counterpart. This behavior indicates a larger acidity of 1-Z gained by the intramolecular hydrogen bond formation.
Conclusions
The solid-state structural investigation trough single-crystal X-ray diffraction reveals a crystal packaging governed by N1־H1 …N3 hydrogen bond, forming inverted dimers along [010] direction. The theoretical calculations carried out at DFT: B3LYP/6-311+G (d, p) level of theory showed good correlation with experimental data, yielding error rates less than 1.4% and 0.9% error for bond lengths and angles. Also, spectroscopic properties were successfully calculated employing ab initio calculations. The UV-Vis spectrum was deconvoluted giving the main electronic transitions from HOMO to LUMO+1 and HOMO-1 to LUMO+5.
In solution, this novel compound exhibited UV radiation-mediated configurational isomerism conversion from 1-E to 1-Z confirmed by 1H-NMR evidence: chemical shift change and new signals appearance owing to system asymmetry. Finally, modified electrochemical behavior induced by configurational changes was observed, supporting the applicability of these type of molecular systems as an electrochemical template.

