











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Fullerenes are a family of molecules that own a highly symmetric closed cage structure, exclusively made up of carbon atoms. Unlike the other allotropic forms of carbon, such as diamond and graphite, which have indefinitely extended structures, fullerenes have an exact molecular weight and behave as a molecule.
These characteristics allow fullerenes to be solubilized in organic solvents and, therefore, to be chemically modified, obtaining many derivatives that, in general, preserve the exceptional physicochemical properties of their precursors [1-3].
Fullerenes behave like poly-olefins that are deficient in electrons; thus, they share the typical reactivity of these type of molecules. Several synthetic strategies were developed to functionalize this class of compounds, including Diels-Alder cycloaddition [4], Bingel-Hirsh cyclopropanation [5], and 1,3-dipolar cycloaddition of azomethine ylides via the Prato reaction [6], [7]. Among all of the previously mentioned methodologies, the Bingel reaction has undoubtedly been one of the most employed approaches to obtain various fullerenes derivatives. This reaction consists of a cyclopropanation that is catalyzed by a strong base, usually sodium hydride (NaH) or 1,8-diazabicyclo (5.4.0) undec-7-ene (DBU), to introduce a-haloester or α-haloketone ligands to two fullerene carbon atoms [8,9].
This reaction occurs through an addition-elimination mechanism. The first step involves the formation of a carbanion by the deprotonation of the active methylene group, which is then added to one of the sp2 carbons of the double bond. This cyclopropanation process was also carried out with malonates in the presence of I2 or CBr4 and a base, generating in situ a-halomalonate and, subsequently, the respective fullerene derivatives [10,11].
As mentioned above, fullerene derivatives, especially those of C60 and C70, have unique physicochemical properties; for this reason, they have been largely investigated for their potential application in a broad variety of technological areas. Fullerenes have been successfully employed in the development of solar cells [12,13] in the medical field [14] as photodynamictherapy sensors [15], as molecularly photoactive devices [16], as magnetic and catalytic materials [17], and as bacterial growth inhibitors [18]. Fullerenes have played an important role in the field of solar cells as their spherical shape generates a system of delocalized n electrons that allows them to behave as good electron acceptors. Additionally, they have a low reduction potential [19].
The modification of perovskite-type solar cells using C60 and C70 derivatives has generated a lot of interest since these structures stand out from other materials thanks to their interesting characteristics such as high absorption coefficients, simple structures, low process temperatures, and lack of complications due to hysteresis. In terms of the application of symmetrically substituted cyclopropanefullerenes in the design of solar devices, Troshin et al. [20] previously showed that the presence of different substituents in the cyclopropane ring could enhance the energy output parameters of photovoltaic devices. A representative example of this application was the work published by Echegoyen et al. [21] in 2016, in which this type of solar cell was modified with the derivatives 2,5-(dimethylester)-C60, fulleropyrrolidine (DMEC60), and its analogue with C70 (DMEC70) to reach PCE of 15.2% and 16.4%, respectively. In 2017, Echegoyen et al. [22] modified these materials using a dimer derivative of C60 (D-C60) formed from two units of the [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM). By doing this, they obtained a cell with the ability to efficiently passivate the energy states between the layers of fullerene and perovskite, which leads to better electron extraction and a photovoltaic performance of 16.6%. Recently, Ye et al. [23] made modifications to the perovskite solar cells (PSCs) by adding the bis(1-[3-(methoxycarbonyl)-propyl]-1-phenyl)-[6,6]C62(bis-PCBM) doped with decamethylcobalt (DMC), which generated a device with both good stability and an efficiency of 20.14%.
This investigation focuses on the synthesis, characterization, and study of the photovoltaic properties of the diacetylmethane monoadducts of C and C These derivatives were obtained from a Bingel reaction, and each of the respective fullerenes, carbon tetrabromide, and DBU in ort/jo-dichlorobenzene (o-DCB) were adopted as shown in Figure 1. This exo-functionalization was carried out to generate a greater structural diversity on C60 and C70 fullerenes and, consequently, to synthetize compounds that can increase the efficiency of energy conversion in perovskite-type solar cells. These compounds have a straightforward, non-expensive synthesis and are easily purified.

Materials and methods
Reagents
(C60 I h ) [5,6] fullerene, (C70-D5h) [5,6] fullerene, 1,8-Diazabicyclo [5.4.0] undec-7-ene (DBU), and carbon tetrabromide (CBr4) were purchased from Sigma Aldrich® (USA); they were used without prior purification. Chloroform (CHCl3), carbon disulfide (CS2), o-dichlorobenzene (o-DCB, C6H5Cl2), and hexane (C6H14) of analytical grade and silica gel (63-200 Mesh) were purchased from Panreac®. Acetylacetone (acac, C5H8O2) was purchased from Alfa Aesar®, and chloroform-d1 (CDCl3) was purchased from Merck®(EE. UU.). The reagents were analytical grade. The TLC conditions were: silica-gel 60 F layers with fluorescence indicator.
Instruments
IR spectra of the products were recorded with a Shimadzu® (Japan) IR prestige-21 spectrophotometer in ATR mode. The spectra were processed with the IR_solution® software.
UV-Vis spectra were recorded on a ThermoScientific® (United Kingdom) Evolution-300 spectrophotometer by dissolving a sample of the solid product (ca. 5 mg) in chloroform. The spectra were recorded between 250 and 750 nm wavelength (X) using a quartz cell (optical path of 1 cm). The spectra were processed using the VisionPro® software.
1H and 13C NMR spectra were recorded on a Bruker® (Germany) 400 MHz spectrometer, using chloroform-d1 (CDCl3) as the solvent and internal standard. The spectra were processed with the MestReNova® software.
Cyclic Voltammetry (CV) and Osteryoung Square Wave Voltammetry (OSWV) were conducted in o-DCB with NBu4PF6 as the supporting electrolyte and using a 1-mm-diameter glassy carbon disk as the working electrode. Ferrocene was added at the end of the experiments to be used as internal reference to measure the potentials.
Matrix-assisted laser desorption/ionization - time of flight - mass spectrometry (MALDI-TOF-MS) was carried on positive mode, with samples dissolved in chloroform and using 1,1,4,4-tetraphenyl-1,3-butadiene (TPB) as the matrix.
Current-Voltage (J-V) performance of the devices was tested using a Keithley 2420 source meter under a Photo Emission Tech SS100 Solar Simulator. The light intensity was calibrated using a single-crystal Si photovoltaic cell as the reference. The measurements were made in air at room temperature.
General methodology for the C60 and C70 cyclopropanation reaction
The C60/C70 fullerene (1 mmol) was dissolved in 5 mL o-dichlorobenzene (o-DCB) in a 25 mL round bottom flask. Then, the acetylacetone (0.5 mmol) and CBr4 (0.5 mmol) were added to this solution. Subsequently, the DBU (0.5 mmol), previously dissolved in 3 mL of the o-DCB, was added drop-by-drop at room temperature to the reaction mixture under stirring. The total time of the reaction was one hour. Once the reaction was finished (TLC check adopting CS2 and CHCl3 as the mobile phase, silica-gel 60 F254 layers with fluorescence indicator, room temperature), the crude reaction product was purified by column chromatography on silica gel (17 g), using hexane, carbon disulfide, and chloroform as eluents. The chromatographic separation for both C60 and C70 derivatives was carried out by first using hexane to extract the o-DCB, then carbon disulfide to separate the unreacted precursor, and finally chloroform to obtain the monoadduct of interest.
Synthesis of (C60-Ih )[5,6]fullerene diacetylmethane monoadduct, (3), C65O2 H6
By employing 100 mg (0.14 mmol) of C60, 7.23 μL (0.07 mmol) of acetylacetone, 23.21 mg (0.07 mmol) of CBr4 and 10.47 pL (0.07 mmol) of DBU, 39.5 mg (0.04 mmol, 69%) of the monoadduct was obtained (3), C65H6O2 (818.65 g/mol) as a brown solid. Rf: 0.62, IR(ATR): v (cm-1): 2920, 2849, 1708, 1646, 1461, 1427; 1H-NMR (400 MHz, CS2/CDCl3): δ (ppm) 2.84 (s,6 H); 13C-NMR (100 MHz, CS2/CDCl3): δ (ppm) 193.44, 144.53, 145.25, 145.10, 144.80, 144.76, 144.61, 143.79, 143.22, 143.11, 142.32, 142.04, 141.26, 137.77, 74.10, 70.37, 28.12; UV-Vis (CHCl3) λ/nm): 680, 425, 488. Expected exact mass: 818.7, experimental m/z: 818.1.
Synthesis of (C70-D5h)[5,6]fullerene diacetylmethane monoadduct, (5), C75O2H6
By employing 100 mg (0.12 mmol) of C70, 6.2 μL (0.06 mmol) of acetylacetone, 19.89 mg (0.06 mmol) of CBr4, and 8.97 μL (0.06 mmol) of DBU, 25 mg (0.02 mmol, 44%) of the monoadduct was obtained (5), C75H6O2 (938.75 g/mol), as a black solid. Rf: 0.56, IR(ATR): v (cm-1) 2917, 2847, 2156, 1707, 1645, 1515; 1H-NMR (400 MHz, CS2/CDCl3): 8 (ppm) 2.80 (s, 6 H); 13C-NMR (100 MHz, CS2/CDCl3): 8 (ppm) 194.59, 155.33, 151.78, 151.15, 150.79, 150.55, 149.39, 149.20, 149.14, 148.53 (2C), 148.51, 148.48, 147.49, 147.35, 146.96, 146.38, 145.94, 145.83, 144.77, 143.99, 143.78, 143.58, 142.78, 141.46, 141.03, 139.71, 136.40, 133.74, 132.86, 130.88, 130.78, 68.24, 67.30, 29.72; UV-Vis (CHCl3) λ/nm: 463, 402, 370, 355. Expected mass: 938.8, experimental m/z: 938.1.
Results and discussion
Monoadducts (3) and (5) were synthesized using Bingel cyclopropanation reaction by combining the selected fullerenes (C60 and C70) with acetylacetone in the presence of CBr4, DBU, and o-DCB as solvent, see Figure 1; o-DCB was chosen as solvent because of the solubility of the precursors in it (C60 27 mg/mL, 36.2 mg/mL C70) [24,25]. The reaction firstly involves the formation of the a-bromoacetylacetone; then it is followed by an in situ cyclopropanation, catalyzed by DBU base, and the product of interest is obtained. DBU is a strong heterocyclic base that reacts rapidly; thus, its addition to the reaction mixture needs to be controlled and slowed to avoid the formation of poly-adducts. The adducts are slightly soluble in chloroform, so it is necessary to add carbon disulfide to have a better result during spectroscopic analysis.
Synthesis of C 60 fullerene diacetylmethane monoadduct
At the beginning of the synthesis of derivative (3) the C60 solution appears violet, but after the addition of DBU the color quickly assumes a wine-red tonality, indicating that a chemical transformation occurred. Employing these working conditions, the product obtained appears to be a brown solid with a 68% yield (42% considering the recovered precursor). It is interesting to note that the TLC (check adopting CS2 and CHCl3 as mobile phase on silica) performed as a control during the synthesis showed the formation of the product of interest (3) and as well the formation of other co-products of greater polarity, which, possibly, correspond to poly-adducts generated within the reaction medium. However, these poly-adducts are practically impossible to avoid, because the presence of 30 double bonds [6,6] in C60 make it difficult to solely obtain the regiospecific derivatives.
It is important to highlight that only two previous works related to the synthesis of this derivative have been reported in the literature. The first is the work done in 1998 by Bingel who obtained this compound from the reaction of acetylacetone in the presence of iodine, DBU, and toluene with a lower yield of 18%, possibly due the poor solubility of C60 in this solvent [26]. The second work, previously cited, was published by Jin et al. and describes the synthesis of this monoadduct starting from a-bromoacetylacetone in the presence of DMSO as solvent and Na2CO3 as base to form the derivative (3) with a yield of 34% [8]. Based on the results obtained from previous published research as well as this research, it can be deduced that, by controlling the stoichiometry, DBU concentration and the reaction time, the derivative (3) can be formed with a good yield.
After having separated and purified the products by column chromatography on silica gel, the structural characterization of the product (3) was carried out by UV-Vis, infrared, nuclear magnetic resonance (1H and 13C) spectroscopy, and mass spectrometry (MALDI).
The UV-Vis spectrum gave the first indication that the formation of C60 monoadduct (3) took place because of the appearance of the absorption bands at 425 nm (Figure 2) that show a behavior similar to the characteristics of a C60 monoadduct spectrum [27]. The absorption band at 324 nm was interpreted as an electronic transition from the pi-bonding orbital to the pi-antibonding orbital (π - π *) of the fullerene unit. Additionally, it is blue-shifted with respect to the C60 band due to the disruption of the sp 2 conjugation when the [6,6] double bond is removed. In toluene, there is a bathochromic shift of about 4.0 nm, which can be correlated to the solvation processes [28].
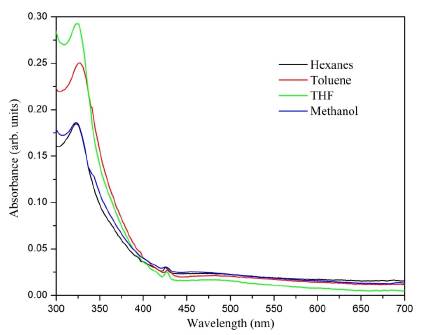
Figure 2 UV-Vis absorption spectra recorded of the compound (3) C60 monoadduct in different solvents.
A secondary indication that confirms the preliminary formation of (3) was found in the infrared spectrum (Figure 3), where the stretching of the sp3 hybridized C-H bond in the methyl group can be observed in the bands at 2920 and 2849 cm-1. Additionally, the absorption bands associated with the out-of-plane and in-plane bending vibrations of the methyl group are observed at 1356 and 1426 cm-1, respectively. Moreover, the absorption band of the C=O stretch was found at 1708 cm-1, while the C=C bond stretch was recorded at 1646 cm-1.

The identity of product (3) was confirmed with the information provided by the nuclear magnetic resonance spectrum. 1H spectrum (Figure 4 a) showed a singlet at 2.84 ppm generated by the methyl protons of the carbon directly bonded to the carbonyl. The 13C spectrum (Figure 4 b) also showed the expected signals. Cycloaddition of a symmetric fragment along the 6,6 bond results in a 13C-NMR spectrum of such a cycloadduct containing 17 peaks of fullerene carbon atoms. Among these, it was possible to observe that σ 70-90 ppm is typical for sp3 hybridized atoms, and the other 16 localized between 137 and 160 ppm are characteristic of the sp2 fullerene carbon atoms.

Figure 4 4. a)1H-NMR spectrum (CS2/CDC13, 60 MHz) and b)13C-NMR spectrum (CS2/CDCl3, 100 MHz) of compound (3) C60 monoadduct. H signals of the monoadduct are singlets. C insets are amplifications of the cage’s chemical shift zone.
Specifically, the signals at 28.12 and 70.37 ppm are characteristic of the sp3 carbons belonging to the methyl group and the quaternary carbon present from the adduct, respectively, while the signal at 74.10 ppm is due to carbons 1 and 2 from the cage. The signal observed at 193.44 ppm, close to the intense CS2 solvent pick, is due to the carbonyl group, and only 13 of the 17 signals expected for the cage (C3-60) are observed since some signals overlap. All signals confirm a C 2v symmetry for the monoadduct, as expected [9]. These results agree with those reported by Bingel [26].
Synthesis of C70 fullerene diacetylmethane monoadduct
A color change occurred during the reaction; in fact, the crude mixture turned from red to dark brown. This indicated that a chemical transformation was taking place. The reaction was followed-up by TLC, where the expected monoadduct product appeared, eluting below the precursor. Similarly to the synthesis of (3), other products, which correspond to poly-adducts formed as a consequence of the multiple reactive bonds present in fullerenes were eluted below (5) in the chromatographic column.
It is important to highlight that C70 has four types of [6,6] bonds, referred to as y, δ, α, and β [27,29]. These last two bonds, in particular, prove to be the most reactive in this molecule, and that is why this reaction can generate both the α and β isomer. However, the literature reports that the Bingel reaction generates a product with high regioselectivity on the C70. This is a consequence of the nucleophilic attack on the α-bond close to the polar zone of the fullerene [30] where the pyramidalization (the distortion from the plane geometry to a pseudo-tetrahedral symmetry) of the sp2 hybridized carbon atoms and the angle of curvature of the fullerene decreases moving from the poles to the equator, and, therefore, the attack on a a-bond becomes the most feasible for a transannular structure, such as the one formed during the Bingel reaction. The reason why this can be assumed with good probability is that the product obtained under these reaction conditions was the a isomer [31,32]. This behavior was also reported with hydroalkylations, hydroarylations, Diels-Alder reaction, and dipolar cycloadditions [3+2] of nitrile oxides in C70. It is worth mentioning that the 1,3-dipolar cycloaddition of azomethine ylides is the least regioselective reaction on C70 since it leads to the α, β, and y isomers in different proportions, depending on the reaction conditions and the nature of the azomethine ilyde [32,33]. It is important to mention that we did not find the β isomer while carrying out column chromatography and purification, and we also did not find it in NMR spectra.
The UV-Vis spectrum (Figure 5) shows four absorption bands recorded at 461, 405, 371, and 352 nm, which appear similar to those reported in the literature for a Bingel monoadduct substituted in the a position of the C70 [34,35]. This suggests that the compound of interest was formed. Similar to (3), each absorption band recorded in the spectrum was associated with possible electronic transitions: The transitions of the carbonyl group from the non-bonding orbital to the pi-antibonding (n- π *) at 461 nm, from the pi-bonding to pi-antibonding (π - π *) at 405 nm, and the transitions of the fullerene double bonds from the pi-bonding to pi-antibonding orbital (π - π *) at 371 and 352 nm. The intensity of the absorption bands decreases as the polarity of the solvent increases, and this can be attributed to the stability of the charge-transfer states in the C70 monoadduct, which is more stable in polar solvents, such as methanol [36]. In the same way as the C60 monoadduct, the C70 presented a brief solvatochromic effect of about 4 nm in toluene, possibly due to the solvation.

Figure 5 UV-Vis absorption spectra of the compound (5) C70 monoadduct, recorded in different solvents.
An IR spectrum analysis allows an initial raw characterization of the product from the cyclopropanation (5). The spectrum (Figure 6) shows the vibration bands corresponding to the symmetric and asymmetric stretching and out-of-plane and in-plane bending of the methyl group in the following regions: 2916 - 2848 cm-1, 1353 cm-1, and 1426 cm-1, respectively. The presence of the carbonyl group is confirmed by the presence of the intense absorption band at 1707 cm-1, caused by the stretching of the C=O bond. The presence of the C=C bond is confirmed by the absorption band at 1645 cm-1, which is caused by its stretching vibration.

The 1H NMR spectrum (Figure 7 a) provided valuable information that confirmed the formation of the product of interest, since the singlet registered at 2.80 ppm corresponds to the six symmetrical methyl protons present in (5). Like the spectrum of the C60 monoadduct, there are other signals that do not belong to the C70 monoadduct that are due to impurities present in the sample, which register between 0.88-1.44 ppm, and could possibly be from grease present in the materials employed. The 13C NMR spectrum (Figure 7 b) reports the expected signals. In particular, the carbons of the methyl group appear at 29.72 ppm, the ring carbon (sp3 -C) at 67.30 ppm, and the carbons 1 and 2 of the cage at 68.24 ppm. The signal of the carbonyl group can be observed at 194.59 ppm, and for the 34 expected signals of the sp2 hybridized carbons from the fullerene cage (C3-60), only 31 signals are observed in the range 130.74 to 155.33 ppm due to peak overlapping. All the signals confirmed the presence of the expected C 1 symmetry for the monoadduct. These results agree with those previously published by Bingel and Hermann et al. [34] and, as previously mentioned, the product obtained appears to be the a derivative due to the bond's very high reactivity [37,38] The formation of other monoadducts was not detected, and, according to the current knowledge, there are no previous reports of the C70 diacetylmethane monoadduct.

Electrochemistry
Previous studies reported that Bingel derivatives of C60 and C70 exhibit quasi and reversible cathodic electrochemical behavior [39-41] CV of C60 diacetylmethane (3) and C70-diacetylmethane (5) compounds were recorded at a scan rate of 100 mV/s (Table 1 and Figure 8). Compound (3) exhibits seven irreversible reduction peaks in which four are well-defined. Based on their shape and current intensity, these are likely to be monoelectronic. On the other hand, compound (5) exhibits six reduction events. Four of these were well-defined, and as with compound (3), all the peaks are irreversible and monoelectronic. No oxidation events were observed for either compound-at least none within the work potential window.

Table 2 Diffusion coefficients for each reduction potential of fullerene acetylacetone monoadducts.

* The molar concentration of each solution was 4.3x10-6 mol/mL.

Figure 8 Cyclic voltammetry (0.05M TBAPF6/o-DCB, 100 mV/s scan rate) of compound (3) C60 monoadduct and compound (5) C70 monoadduct. Dotted lines are the individual scans for each peak potential.
Figure 9 shows the cyclic voltammogram for the first reduction potential of compound (3) (left figure) and its behavior as the scan rate is increased from 100 to 500 mV/s. The current intensity is increased, and the potential is shifted as the scan rate goes higher, which is a typical characteristic of irreversible processes. This behavior was also observed for compound (5) (right figure). Furthermore, there is a linear dependence between the peak current and the square root of the scan rate, a fact that confirms that the electron transfer is regulated by diffusion. With this knowledge, it is possible to calculate the diffusion coefficients through the Randless-Sevcik equation (see Figure 10 and Table 2).

Figure 9 Cyclic voltammetry of the first peak reduction at different scan rates for a) compound (3) C60 monoadduct and b) compound (5) C70 monoadduct.

Figure 10 Dependence of the current peak with the square of the scan rate for each reduction potential of a) compound (3) C60 monoadduct and b) compound (5) C70 monoadduct.
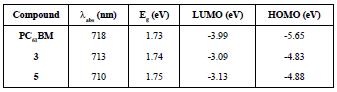
Furthermore, Osteryoung Square Wave Voltammetry (OSWV) was carried out for compounds (3) and (5). For both compounds, six reduction waves were observed (Table 3 and Figure 11). Both fullerene derivatives exhibit a cathodic shift in the first reduction potential, compared to the pristine C60 fullerene and C70 fullerene. Based on the first reduction potential, the LUMO energy level was calculated to be -3.09 and -3.13 eV for (3) and (5), respectively. Comparing these values with PC61BM (-3.90 eV) and other Bingel fullerene derivatives, the compounds of this study are also higher than the LUMO energy of the traditional perovskite MAPbI3, which allows the conductance of the electron flux.
Table 3. Comparison of the potential reduction values of fullerene acetylacetone monoadducts and pristine fullerenes.

aFrom the reference[42] b From the reference[43] c From the equation LUMO = -(4.72 + Ered on).

Figure 11 Osteryoung Square Wave Voltammetry in 0.05M TBAPF6/o-DCB of compound (3) C60 monoadduct and compound (5) C70 monoadduct.
It is possible to replace C60 with C70 in derivatives for the electron transport layer (ETL) and develop highly efficient solar cells [44]. The loss of symmetry in the fullerene C70 makes fewer forbidden transitions, representing a higher probability for HOMO-LUMO transitions [45]. As we can see in the UV spectra (Figure 2 and Figure 5) and the cyclic voltammetry (Figure 9), the differences in the spectral range of absorption between both derivatives allows us to conclude the highest efficiency value is expected for compound (5). As expected, there is a significantly stronger absorption in the visible region, and an increase in the external quantum efficiency for C70 adducts.
According to Dai and co-workers, the different molecular aggregation can explain the results of a better ETL by using a formulation with a mixture of the PC71BM isomers instead the isolated isomers [46]. This fact, and the higher solubility of C70 compounds compared to C60 ones, reduce the disorder in the ETL, increasing the quasi-Fermi level of the photogenerated electrons and also reducing the pinholes as well [47]. This aspect can be confirmed with the higher value of Voc and may also elucidate the different values of PCEs in this work.
Photovoltaic Performance
The results from the electrochemistry combined with the UV-Vis characteristics allowed us to obtain the work function for the compounds (3) and (5), which is vital information to determine whether they are suitable or not to be used as an ETL in PSCs. HOMO and LUMO values of compounds (3) and (5) are summarized in Table 4. Figure 12 shows that HOMO-LUMO values of the compounds (3) and (5) do not match very well with those of the perovskite layer, which can lead to an inefficient hole blocking process and hysteresis behavior in the PSCs. This phenomenon makes part of the experimental work in the understanding of charge generation at organic heterojunctions and charge separation. High-level theoretical calculations as semiempirical and density functional molecular calculations could be carried out in order to understand the dynamics of charge pair generation and correlate the theoretical LUMOs with the open circuit voltage of the photovoltaic device [48,49].
Table 4 Photovoltaic performance of the control PC61BM, of compound (3) C60 monoadduct and compound (5) C70 monoadduct.

Device fabrication
The device fabrication followed the procedure adopted by Fernández-Delgado et al. [50] who probed the variation of interfacial interactions of PC61BM-like electron transporting compounds for PSC.
PSCs with an ITO/NiOx/CH3NH3PbI3/ETL/Ag configuration were fabricated (Figure 12) to study the capacity of the compounds (3) and (5) to act as ETLs. Chlorobenzene was employed as a solvent for the deposition of adducts (3) and (5).
The J-V curves for the fabricated PSCs are shown in Figure 13. PC61BM as ETL was used as the control and revealed an average PCE value of 14.9%, an open circuit voltage (Voc) of 1.01 V, a fill factor (FF) of 64%, and a short circuit current (Jsc) of 23.0 mA/cm2. The PSCs performances are summarized in Table 5.

Table 5. Photovoltaic performance of the control PC61BM, of compound (3) C60 monoadduct and compound (5) C70 monoadduct.

The results show a clear difference between the two fullerene derivatives, with compound (5) showing a better performance with values of PCE very close to that of the control device. As can be distinguished in Figure 13, compound (5), has a better performance, a smaller hysteresis, and a higher current density and voltage value. This behavior can be explained by a better matching of the energy levels to those with the perovskite by an enhanced light absorption of this material and better interface perovskite/ fullerene quality, which results in less hysteresis, better FF, and reduced current leakage. All those factors can improve device performances, making this material a suitable ETL in PSCs.
Conclusions
By controlling the stoichiometry and the reaction time, and through a knowledge of the mechanistic issues of the fullerene functionalization reactions and their complex molecular geometry that differentiates their reactivity, the synthetic yield of the C60 diacetylmethane monoadduct was improved, with respect to what was previously reported in the literature. In addition, the new compound, the C70 diacetylmethane monoadduct, was synthesized, and it was obtained with a moderately high yield. The monoadducts were tested in photovoltaic performance studies, and a solar cell efficiency of 8.5 and 14.0 was obtained for compound (3) and (5), respectively. The low values are related to the mismatch in the energy levels of the derivatives when compared to the perovskite, which produces hysteresis and current leakage in the PSCs. Even though the compounds were not very compatible as an ETL, compound (5) exhibited the highest performance, very close to PC61BM, which was used as a control. Likewise, OSWV and CV were conducted for compounds (3) and (5), showing irreversible reduction potentials controlled by electron transfer diffusion. Both compounds present LUMOs energy levels higher than the traditional MAPbI3 perovskite one. The optical absorption range and the morphology of ETL may explain a higher value for the C70 derivative compared with C60 diacetylmethane.

