











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis (MBT), remains the most dangerous infectious disease globally. Every day 30,000 people contract this disease, and more than 4,000 people die. The large number of people infected with the human immunodeficiency virus has caused a rapid increase in the prevalence and mortality of TB [1]. However, since 1980, another cause of an increase in TB cases has been resistance to drugs, resulting in the appearance of multidrug-resistant tuberculosis (MDR-TB) and extremely drug-resistant tuberculosis (XDR-TB) [2], thus leaving many patients with no treatment options.
Some treatment procedures for tuberculosis or other diseases consist of building different molecular scaffolds with broad-spectrum biological activity. In other words, molecular hybridisation has provided additional tools for combining various molecular moieties of known molecules to synthesise new compounds that could increase their biological activity. To combat this disease, first-line prodrugs, such as isoniazid (INH) and pyrazinamide (PZA), are used in combination with other first- and second-line drugs (rifampicin, ethambutol, and streptomycin) [3], [4]. INH is one of the most widely used antituberculosis prodrugs. To exert its antimycobacterial effect, INH requires activation by a catalase-peroxidase called KatG to form an isonicotinoyl radical. This radical binds to nicotinamide adenine dinucleotide (NAD+), and the resulting adduct inhibits enoyl-acyl carrier protein reductase (InhA), which is responsible for the biosynthesis of mycolic acids in the bacterial cell wall [5], [6]. Similarly, prodrug PZA must be converted to its active form, pyrazine acid (POA), to exert its antimycobacterial effect. Upon POA accumulation, the internal compartment acidifies, thereby disrupting the function of the mycobacterial cell wall [7], [8].
The emergence of drug-resistant strains represents a significant problem for public health, and the administration of multiple drugs requires excessive compliance and commitment by the patient. Hence, it is essential to develop and evaluate derivatives or hybrid molecules capable of modulating various targets to provide a broader range of pharmacological agents [9]. New and more biologically-potent antituberculosis compounds derived from PZA [10], [11], INH derivatives [12], [13], and INH and PZA hybrids have been reported, along with other drugs such as POA [14], quinolone [15], phenanthroline [16], benzofuran [17], 4-phenylthiazol-2-amine [18], oxadiazole [19], and piperazine ferulate [20], among others. These compounds have greater antituberculosis and antibacterial properties than free drugs because hydrazone residue-containing compounds exhibit potent photophysical bioactivity [21].
A promising approach in the search for new drugs for the treatment of tuberculosis involves the use of hybrid compounds where two or more drugs are combined in a single molecule; these compounds can offer an extra benefit, mainly by claiming to enhance and reduce toxicity. Some examples of antituberculous hybrids are isatin-thiozole [22], bequinolone-isoanizid [23] and benzofuran-oxadiazole [24] Therefore, we replicated the synthesis of a PZA and INH hybrid bound by the Schiff base group [25]. This hybrid can be considered a prodrug for TB treatment and can act synergistically. This report describes the details of ultrasound-assisted synthesis optimisation, which minimised reaction time and improved performance. The antituberculosis activity of the hybrid was assessed using the Mycobacterium tuberculosis (MBT) H 37Rv strain, obtaining a lower minimum inhibitory concentration (MIC) compared with that obtained using the free drugs (PZA and INH).
Materials and methods
All chemicals were purchased from Sigma-Aldrich, St. Louis, Missouri, USA. Spectroscopic grade solvents were used without further purification. Compound synthesis was performed using a 40-kHz, 120-W ARS-XQXJ-002H ultrasonic cleaner with a 3-L tank. Infrared spectra were recorded with KBr pellets ranging from 4000 to 400 cm-1 on a Shimadzu FT-IR Prestige 21 spectrometer. The 1H nuclear magnetic resonance (NMR), 13C NMR, and Heteronuclear Single Quantum Coherence (HSQC) spectra were obtained using a 500-MHz BRUKER Ascend spectrometer in a dimethyl sulfoxide-D6 (DMSO-d6) solution with tetramethylsilane as an internal standard. Elemental analysis was performed using a CE-440 elemental analyser, EAI Exeter Analytical Inc. Melting points were determined using a Thermofisher Scientific Electrothermal IA9100 melting point apparatus (USA). The ultraviolet-visible spectra were recorded in the range of 200400 nm on a Shimadzu UV 1800 UV-Vis spectrometer (Japan) using a 1-cm quartz cuvette at room temperature (RT).
MBT H37Rv isolates were obtained from Cayetano Heredia University Hospital. The suspensions were prepared in 10% Tween 80 (v/v) (Sigma Chemical Co., St. Louis, Mo., USA) in such a way that their degree of turbidity coincided with McFarland Standard No. 1 (approximately three x 107 CFU/mL) [25]-[27].
Synthesis optimisation of N-(dimethylaminomethylene) pyrazine-2-carboxamide (1)

A mixture of PZA (123 mg, one mmol) and N,N-dimethylformamide dimethyl acetal (400 µL, three mmol) was sonicated for 30 min at room temperature. N,N-dimethylformamide dimethyl acetal (200 µL, 1.5 mmol) was added, followed by sonication for 30 min at room temperature. (Note: Ice was added to the water bath after 30 min as it was an exothermic reaction). After the reaction was complete, the solid product in suspension was separated from the liquid by filtration and washed three times with diethyl ether in the ultrasonic cleaner (3 x 40 s). The product was then recrystallised with acetonitrile at 50 °C, filtered, and vacuum dried.
White solid (1), 75% yield; (m.p.) 192 °C. Lit. [28] m.p. 192-193 °C. IR (KBr): 3047, 3010, 2927, 2929, 2812, 1651, 1614, 1566, 1483, 1421, 1337, 1265, 1253, 1161, 1112, 1016, 1047, 920, 786, 624 cm−1. 1H NMR (500 MHz, DMSO-d6) δ9.34 (s, 1H), 8.74 (s, 2H), 8.68 (s, 1H), 3.21 (s, 3H), 3.15 (s, 4H).13C NMR (125 MHz, DMSO-d6) δ 35.76, 41.62, 144.88, 146.22, 146.76, 149.59, 161.87, 174.01. Anal. Calc: C8H10N4O (178.25): C, 53.80; H, 5.39; N, 31.19. Found: C, 53.92; H, 5.66; N, 31.44.
Synthesis optimisation of the hybrid N-[(2-iso-nicotinoyl hydrazine) methylidene-2-pyrazine carboxa mide] (2)

To obtain compound 2, compound 1 was reacted with INH using pyridinium p-toluenesulfonate as a catalyst. N-(dimethylaminomethylene) pyrazine-2-carboxamide (1) (500 mg, 2.25 mmol) was sonicated with acetonitrile (15 mL) for 15 min until the product was dissolved at 60 °C in an ultrasound bath. The pyridinium p-toluenesulfonate (743 mg, 2.25 mmol) catalyst was added at a temperature of 60 °C and sonicated for 15 min until dissolved. After 15 min, the INH solution (310 mg, 2.25 mmol) dissolved in acetonitrile (35 mL) was added at a temperature of 60 °C (previously stirred separately). The mixture was stirred at 600 rpm for 45 min at the same temperature and then cooled. Subsequently, the solid was separated by filtration and vacuum dried. The white solid obtained was extracted using methanol (5 x 50 mL) and concentrated solvent. Lastly, the resulting hybrid was washed using ethanol to remove impurities and vacuum dried at RT.
White solid (2), 70% yield; (m.p.) 230.5 °C. Lit. [28] m.p. 230-232 °C. IR (KBr): 3263, 3207, 3093, 3057, 1687, 1654, 1620, 1548, 1413, 1303, 1284, 1228, 1176, 1130, 1070, 1022, 925, 848, 740, 684 cm−1.1H NMR (500 MHz, DMSO-d6): δ 11.76 (s, 1H), 11.42 (s, 1H), 9.26 (d, J = 1.5 Hz, 1H), 9.07 (s, 1H), 8.94 (d, J = 2.45 Hz, 1H), 8.81 (dd, 1H), 8.78 (d, J = 1.65 Hz,2H), 7.82 (d, 2H). 13C NMR (125 MHz, DMSO-d6): δ 163.6, 161.5, 150.7, 149.9, 148.7, 144.6, 144.3, 142.1, 140.9, 121.8. Anal. Calc: C12H10N6O2 (270.25): C, 52.90; H, 3.61; N, 30.81. Found: C, 53.33; H, 3.73; N, 31.10.
Evaluation of biological activity
The PZA susceptibility test for the MBT H37Rv strain was performed at a Level III biosafety laboratory using the tetrazolium microplate assay for cell viability (TEMA) [3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] (Aldrich Chemical Co., Milwaukee, Wis., USA), which is based on the colour change to blue using tetrazolium bromide, indicating active cellular respiration [29]-[31].
With this technique, MICs of the drugs were determined by means of a series of 1:2 dilutions in a 96-well microplate at pH 6.0 and 6.7, with concentrations starting at 6.5 mM for PZA, INH, and compound 2 in 7H9 medium. INH was evaluated in the microplate at pH 6.0, with the INH concentration starting at 0.026 mM. The PZA, INH, and compound 2 solutions were sterilised by filtration and mixed with a 7H9 medium. We also established control for monitoring MTB culture without the drugs.
The MTB H37Rv strain was used because this strain is a wild-type strain naturally sensitive to PZA. The MTB suspension was prepared from a primary culture in a 7H10 agar medium and incubated for 2-3 weeks. Bacterial m bass was harvested in a glass tube with 100 µL 0.1 % (v/v) Tween 80 and 3-mm glass beads. After vortex homogenisation, turbidity was adjusted to the McFarland No. 1 standard (3 x 108 CFU/mL) using a 7H9 medium. Finally, McFarland #1 strain was diluted in 7H9 in a ratio of 1:25 [14]. In each well, 100 µL of 1:25 MTB suspension was transferred. The tests were performed in duplicate for each concentration of the drugs. The microplates were placed in Ziploc bags and incubated at 37 °C for six days.
Results and discussion
Synthesis
Ultrasound-assisted organic synthesis, which has garnered increased attention in recent years [32], continues to be a challenge and a topic of great interest. The use of ultrasound, a new method free of solvents and secondary reactions, improves reaction rates and decreases residue compared to traditional techniques such as agitation at constant temperature or under conventional heating conditions [33]. A reaction time of 7 h is required to obtain the reported compound 1 [28]. For this reason, ultrasound was used to optimise synthesis as it involves more excellent compound dispersion [34], requires a shorter reaction time (1 h), increases the yield to 95% without recrystallisation, and results in 30% more output from the recrystallised product as compared to the previously reported synthesis method. The hybrid or compound 2 was obtained by modifying the reported process: reducing the chemical synthesis time to 1 h at a temperature of 60 °C in the ultrasound bath. The final product (2) was obtained at a 70% yield and was soluble in water, methanol, dimethyl sulfoxide, and dimethylformamide.

Spectroscopic measurements for compound 1
The absorption spectrum of the PZA derivative (1) presents two bands. The first band at 210.5 nm (π→π* transition) corresponds to the carbonyl group, whereas the second is present at 291.8 nm (n→π* transition). The infrared spectrum of compound 1 presented bands at 3,047 and 3,010 cm-1 corresponding to the v(C-H) stretching of the aromatic ring; the bands at 2,929 and 2,972 cm-1 are due to v(CH3) testing of the hydrogen bonds; and the intense band at 1,651 cm-1 belongs to the tertiary amide (N-C=O). In addition, the band at 1,614 cm-1 reflects the formation of the Schiff base (N=C)[35], and the bands of compound 1 around 1,265 and 1,253 cm-1 are assigned to the (C-N) bond.
The 1H NMR spectrum of compound 1 in solution (DMSO-d6) shows five signals; the most shielded signals, found at 3.15 ppm (CH3) and 3.21 ppm (CH3), correspond to -CH3 protons of the tertiary amine. The signals obtained in the less shielded regions at 8.73 (H5 and H6) and 9.34 ppm (H3) correspond to the pyrazine ring. The signal observed at 8.68 ppm (CH) corresponds to the Schiff base (N=C). The 13C NMR spectrum of compound 1 in solution (DMSO-d6) presented eight carbon signals: 35.31 and 41.16 (CH3) ppm, 144.44 (C3) ppm, 145.78 (C5) ppm, 146.32 (C6) pm, 149.15 (C2) ppm, 161.43 ppm (N=C), and 173.57 (C=O) ppm, whose assignments for each carbon atom were verified and compared with the results obtained by Imramovsky [28].
Spectroscopic measurements for compound 2
The absorption spectrum of the hybrid (2) presents three bands. The first band at 214 nm (π →π* transition) corresponds to the carbonyl group (C=O). In contrast, the second and third bands are observed at 278 nm (n→π * transition) and 302 nm (n→π * transition), indicating that the displacements of the last two bands belong to the aromatic groups of INH and PZA, respectively. The infrared spectrum of the hybrid (2) presents two broad bands characteristic of the NH group at 3,262 and 3,207 cm-1. Moreover, it shows bands at 3,093; 3,072; and 3,057 cm-1 representing the aromatic ring's v(C-H) stretching. Additionally, there are three leading bands: the first corresponds to the carbonyl group of INH (1,687 cm-1), the second corresponds to the carbonyl group of PZA (1,651 cm-1), and the third corresponds to the secondary amide of INH, which corresponds to a range of 3,100-3,400 cm-1 [35], [36].
The 1H NMR and 13C NMR spectra of the hybrid and 1H {13C} NMR/ HSQC two-dimensional spectrum enabled the assignments of all carbon and hydrogen atoms.
The 1H NMR spectrum (Table 1) shows the presence of eight signals corresponding to protons present in compound 2, whose assignments were reported to be as follows [28]: 7.82 (m, 2H), 8.80 (m, 3H), 8.94 (d, J = 2.4 Hz, 1H), 9.08 (br s,1H), 9.27 (s, 1H), 11.39 (br s, 1H), and 11.75 (br s, 1H).
In Figure 4, the signals obtained in the less shielded regions are protons corresponding to the amino groups (N-H), where the sign of the N-H attached to the carbonyl group (C=O) presents more significant displacement because it is surrounded by a more electronegative environment.

The H3, H6, and H5 signals correspond to the aromatic ring of pyrazine, where the sign of the H3 proton signifies that it is the most displaced proton of the ring. This is due to its closer proximity to the carbonyl, also observed as a doublet at 9.26 ppm (1H) because it is coupled to the H5 proton with J3-5 = 1.5 Hz. The pair observed at 8.94 ppm (1H) represents the H6 proton coupled to the H5 proton with J6-5 = 2.45 Hz.
Likewise, the signal of the H5 proton is observed at 8.81 ppm as a double doublet because it is coupled to the H6 proton with J = 2.45 Hz (J5-6 = J6-5) and the H3 proton with a J5-3 = 1.5 Hz.


The aromatic signals of the pyridine ring show less displacement as compared to those of the pyrazine ring, where the movements of the H8 and H12 protons are observed in the less shielded regions (7.82 ppm). This is due to their more excellent proximity to the heteroatom of the pyridine ring, which causes greater displacement owing to the high electronegativity of its nitrogen atom. The signals of the H8 and H12 protons are equivalent and appear as a doublet coupled to H9,11 (J = 6 Hz). The signs of the H9 and H11 protons are also equivalent and appear at 8.78 ppm as a doublet and are coupled to H8,12 (J = 6 Hz).
The 13C NMR spectrum (Table 2) of compound 2 was obtained in DMSO-d6, where the solvent signal is displayed at 40.08 ppm. Likewise, ten signals correspond to the 12 carbons present in the hybrid structure. The C8 and C12 carbons are equivalent, as are the C9 and C11 carbons. The 13C NMR spectrum in solution (DMSO-ds) of the hybrid has been reported previously [28], where the carbon signals were registered as follows: 109.1, 121.3, 140.4, 141.7, 143.7, 144.1, 148.2, 150.3, 160.9, and 163.0 ppm.

The carbon signals observed in the less shielded regions are carbonyl groups (C=O). This is because they are directly attached to the highly harmful oxygen, leaving the carbon atom unprotected, which results in more significant displacement. The carbonyl group near the pyrazine ring is observed at 163.58 ppm [37], [38], and the carbonyl carbon near the pyridine ring is observed at 161.46 ppm [39].
The C9 and C11 carbons are equivalent; therefore, only one intense signal is observed at 150.78 ppm. The more significant displacement compared to the pyrazine ring carbons is because those carbons are directly bound to the nitrogen heteroatom of the pyridine ring.
The other four carbon signals with the greatest displacement are those of the pyrazine ring due to the resonance of the aromatic ring where the carbons are directly attached to the nitrogen. Another atom with greater electronegativity than carbon has signals situated in C3 and C5 that are observed as a very close signal, indicating that they are equivalent. However, when the spectrum is expanded, separation of the signals can be observed at 144.74 and 144.63 ppm for the C3 and C5 carbons, in that order, with the C3 carbon showing greater displacement due to it is closer proximity to the carbonyl group.
Signals from the other two carbons of the pyrazine ring, the C5 and C6 carbons, are present at 144.26 and 148.74 ppm, respectively. Likewise, the carbon signal that provides evidence of the formation of the hydrazone group (C=N) can be observed at 142.18 ppm [39]. This displacement in the aromatic region is because the hydrazone group is directly attached to the nitrogen (an atom with greater electronegativity than carbon).
In contrast, regarding the other remaining carbons of the pyridine ring, the signal for the C7 carbon can be observed at 140.91 ppm; this more significant displacement than the C8 and C12 carbons is due to the proximity of the C7 carbon to the carbonyl group (C=O). The C8 and C12 carbons are equivalent, and their signal appears at 121.82 ppm as a single peak.
Finally, the HSQC spectrum of compound 2 (Figure 6), which did not show the C2 and C7 carbon atoms and the two carbonyl groups (C=O), indicates that these carbon atoms were not protonated. Despite this, six correlation contours were observed in the spectrum as expected. The carbon atoms in compound 2 are listed as follows: C3, HC=N, C5, C6, C9, C11, and C8, C12; this confirms that they are carbon atoms, each attached to its corresponding hydrogen atom.

Their correlation contours were corroborated with 1H and 13C NMR for the assignment of each proton directly bound to the carbon, thereby enabling complete elucidation of the hybrid by NMR. The correlation of the molecule is shown in Figure 7.

Antimicrobial evaluation of compound 2
The MIC of compound 2, INH, and PZA, the last two being control drugs in the analysis using MTB H37Rv, were determined at pH 6.0 and 6.7. Compound 2 and INH reduced MTB multiplication to 0.025 mM at pH 6.7, and no colour change to blue was observed, indicating active cellular respiration. At pH 6.0, the MIC of compound 2 was also 0.025 mM; however, it should be noted that this concentration completely inhibited the multiplication of MTB H37Rv (Figure 8). In contrast, the MIC of INH at pH 6.0 was 0.013 mM (Figure 8), resulting in a reduction in the mycobacterial load. In contrast, the MIC of PZA on MTB H37Rv was 0.8 mM and 0.4 mM at pH 6.7 and pH 6.0, respectively, by means of TEMA and the observed mycobacterial load (Figure 8).
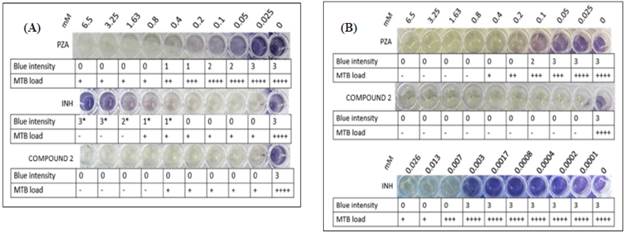
*Compound 2: hybrid; INH: isoniazid; PZA: pyrazinamide; MTB: Mycobacterium tuberculosis.
Figure 8 Determination of the mínimum inhibitory concentration (MIC) of PZA, INH, and compound 2 at pH 6.7 (A) and pH 6.0 (B) on MTB H37Rv by tetrazolium microplate assay and microscopic evaluation. The blue intensity was categorised with 0, 1, 2, and 3; 0 indicates no colour change, 1 and 2 indícate low-intensity colour change, and 3 shows a high-intensity colour change. The MTB load was represented by a plus sign (+) and the absence of MTB by a minus (-) sign.
In Figure 8, it was observed that INH (pH 6.7), at concentrations higher than 0.4 mm, results in a colour change to blue. However, the observed colour change was not due to the mycobacterial load because a plus sign (+) was recorded for the presence and a minus sign (-) for the absence of MTB in these wells.
Acidic pH favours the inhibition of MTB growth. PZA is a first-line antituberculosis drug; its mechanism of action comprises acidifying the intracellular medium of MTB followed by the hydrolysis of PZA to POA by way of the enzyme pyrazinamide. On the other hand, INH induces the production of reactive species, thereby enabling the inhibition of biosynthetic enzymes involved in the synthesis of lipids and nucleic acids [40], [41]. Peterson et ai. (2015) reported that the INH mechanism of action is not influenced by acidic or basic pH [42]. Nevertheless, in this study, we were able to determine that, at an acidic pH (pH = 6.0), the antimycobacterial activity of PZA was relatively favourable compared to that of INH and that pH did not influence the antimycobacterial activity of PZA. However, PZA reduced the metabolic activity of MTB at pH 6.7 without affecting its proliferation; the same effect was observed with 0.007 mM INH at pH 6.0. In contrast, compound 2 showed a better response than PZA and INH. It was determined that its MIC was equal to that of INH and, at a lower concentration, to that of PZA, thereby affecting cell proliferation metabolic activity of MTB. However, it should be noted that lower concentrations of compound 2 were not used, which would be of interest in determining if the MIC is less than 0.025 mM.
Conclusions
The synthesis of compounds 1 and 2 was achieved by way of an ultrasound-assisted reaction and solvent-free procedures (environmentally friendly conditions), which minimised the reaction time to 1 h and resulted in 75% and 60% yields. The chemical structures were elucidated using 1H NMR, 13C NMR, and HSQC, and the results corroborated the spectroscopic data reported in the literature [28]. According to the results obtained by TEMA, the hybrid (compound 2) presented a MIC of 0.025 mM at pH 6.0 and 6.7 against MBT H37Rv. The MIC values at pH 6.7 obtained for the combination (compound 2) provide an equal and better perspective of its potential activity in comparison to INH (MIC = 0.025 mM) and PZA (MIC = 0.8 mM). Nevertheless, both compounds exhibit less activity than INH at pH 6.0.

