











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Parasitic infections remain one of the most pressing global health concerns of our day, affecting billions of people and producing unsustainable economic burdens [1]-[3]. One of these parasites is Leishmania. Leishmania are unicellular parasites that cause human and animal illness, considered as a neglected tropical disease, which is transmitted by infected mosquitoes, resulting in a wide range of pathologies [4]-[6]. Leishmania (L.) donovani, is a species of this group of parasites that causes highly virulent fatal visceral leishmaniasis.
Worldwide L. donovani is known to cause between 50 000 to 90 000 new cases of kala-azar or visceral leishmaniasis (VL) [7]-[9], with a total of ~0.2 to 0.4 million people in 98 countries. Added to this, the World Health Organisation (WHO) considers leishmaniasis to be prominent among the global causes of death by infectious diseases. Due to the global health problems that this parasite entails, the re-evaluation of new chemotherapeutic treatments is necessary, since it is known that the current ones can fail. It is currently known that antimonials are the first line of treatment against leishmaniasis in many regions of the world [10]-[12].
The recurrent use of antimonials and other pharmacological treatments as a treatment for leishmaniasis, have generated resistance of these parasites to these drugs [13]-[15]. This creates a serious problem, especially in regions where sanitary conditions are poor, and the combination of drugs is not feasible. Therefore, the search for new strategies and targets for drug development becomes an imperative need [16]-[18].
In this regard, it is known that proteins related to transcriptional processes could emerge as excellent targets. A group of proteins known as molecular "readers" of lysine acetylation, containing a bromodomain (BRD) have emerged as key gene expression regulators and a promising new class of drug targets ([1], [19], [20]). The bromodomain was first described in the characterisation of the Drosophila melanogaster nuclear remodelling comprised of ~110 amino acids. The bromodomain folds into a left-handed bundle of four a-helices linked by two variable-loop regions (BC and ZA) that form a hydrophobic pocket for the acetylated lysine ([4], [21], [22]).
There is evidence that lysine acetylation is critical in kinetoplastids (Trypanosoma spp. and Leishmania spp.) [1], [23]-[25]. Therefore, in this research we study the dynamics and energetics of the bromodomain of L. donovani in complex with bromosporine, which is considered as a bromodomain pan-inhibitor, with the aim of understanding the mechanism of molecular recognition, and energetics, in order to use this information in future drug development rationally for this group of proteins.
Materials and Methods System setup
The system was prepared by means of the CHARMM-GUI [26] tool, using the Solution Builder [27] module. Missing atoms in the middle of the bromodomain chain (PDBID: 5C4Q) were added with PDBReader [28], the protein was protonated to pH 7.0 with the PDB2PQR tool [29]. Next, the system was solvated with the TIP3P [30] water model, into a 15 Å cubic box of padding, and a 150 mM concentration of NaCl. Finally, the bromosporine inhibitor was pa-rameterised with the CGenff tool [31]. All simulations were run with NAMD 2.14 [32] and CHARMM36 FF [33] using CGenFF [31] for the ligand only.
MD protocol
The constructed systems were minimised in the presence of a harmonic restraint of 5 kcal mol-1 Å-2 on the heavy atoms of the ligand and the backbones of the protein for 10 000 steps. Next, the system was equilibrated in a NVT assembly at a temperature of 300 K for 5 ns, then the system was equilibrated in a second phase in a NpT assembly maintaining a pressure of 1 bar for 10 ns with the same restraint. Finally, production in NpT at 310 K and 1 bar of pressure was performed for 300 ns, for a total of three independent replicates (3 x 300 ns) accumulating a total of 0.9 μs.
In the equilibration and production steps the Lavengin thermostat and the Nosé-Hoover Langevin piston were used to control temperature and pressure. Atoms of rigid hydrogens were maintained with SETTLE algorithm [34] in waters, while to solute the RATTLE/SHAKE algorithm was used [35]. Short-range, non-bonded and long-range non-bonded interactions were treated with a cut-off scheme at 12 Å and PME [36], respectively. The r-RESPA multiple time step scheme [37] was used in all cases with 2 fs time integration steps.
Non-equilibrium free energy calculation
For this, the system was prepared and equilibrated with the standard protocol of QwikMD [38], with the only exception that a 40 Å extra padding was added into the Z axis to enable pulling. For pulling a potential bias of the form
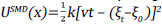 was added for the original potential
was added for the original potential
 through the colvars module [39], getting a modified potential (H
±). For this, the centre of mass of the ligand ξ
L
, was pulling with a speed vof 10 Å ns-1 and к of 7 kcal mol1 Å 2 during 4 ns, enough to reach the unbound state. During the pulling the C
α
atoms were restrained with a force of 5 kcal mol-1 Å -2. A total of 20 independent replicates were performed (total 80 ns). The work was computed for numerical integration using W= v∫0
t1
dt'F(t').
through the colvars module [39], getting a modified potential (H
±). For this, the centre of mass of the ligand ξ
L
, was pulling with a speed vof 10 Å ns-1 and к of 7 kcal mol1 Å 2 during 4 ns, enough to reach the unbound state. During the pulling the C
α
atoms were restrained with a force of 5 kcal mol-1 Å -2. A total of 20 independent replicates were performed (total 80 ns). The work was computed for numerical integration using W= v∫0
t1
dt'F(t').
Based on these simulations, the potential of mean force (PMF(ξ),Φ(ξ) was reconstructed by means of Jarnzynski identity as show in the following equations [40]:
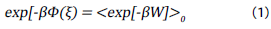
If rearranging the equation, it shields the Ф(ξ):
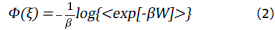
and expanding this equation for the 2nd order cumulant:
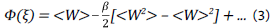
Where Ф(ξ) is the potential of mean force, i.e the free energy profile over the collective variable (ξ), Wis the work done along ξ and β is the inverse of thermal energy (1/k B T),where k B is Boltzmann's constant.
On the other hand, to calculate the ΔG o bind standard, we used the following expression:

then:
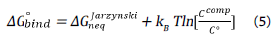
Where ΔG o bind in kcal mol-1, k B in kcal mol-1 К1,Ссошр=1 ligand Box-1 is the concentration used in the simulation in Å3 and C°=1/1661 Å-3 is the standard concentration yielding 1 ligand x 1661 Å 3. These last two terms are added to pass from the computational system to the standard system. Here Ф(ξ) bound and Ф(ξ) unbound correspond to Ф(ξ) the associated and dissociated thermodynamics state (i.e macrostates) respectively.
Molecular data analysis
All analyses were performed through in-house Tcl/Python scripts based on VMD 1.9.4 [41] and MDAnalysis 2.0 [42], respectively. The images were rendered with VMD 1.9.4 [41]. The plots were generated with R 3.4 [43] and the ggplot [44] library. Interactions throughout the simulations were computed with an in-house python implementation of molecular dynamics interaction plot tool (https://github.com/tavolivos/Molecular-dynamics-Interaction-plot).
Results
This work explores the interactions and recognition mechanism of bromosporin in complex with bromodomain. Figure 1 shows the two systems that were constructed, one system (1A) focused on performing conventional molecular dynamics and the second (1B) used to perform the steered molecular dynamics method.
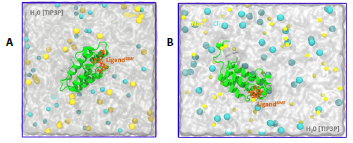
Figure 1 System setup representation A. Conventional molecular dynamics representation and B. SMD of the bromosporine ligand bound to the bromodomain. All systems contain Na* (sky blue) and Cl- (yellow) ions, while water is represented as the surface in white.
Bromodomain contains flexible regions
In order to evaluate changes in the rigidity/flexibility of the bromodomain structure, root-mean-square fluctuations (RMSF) analyses of the backbone were performed. Protein flexibility and dynamics are essential for the molecular recognition process and play a fundamental role in virtually all biochemical processes in living organisms [45]. The results in Figure 2A show that all the replicates present three flexible regions in the protein and present RMSF values from 1 to 4 Å. Furthermore, Figures 2A, 2B and 2C show the structure of the receptor and the location of the flexible regions. R'1 ranges from PRO-36 to THR-50, R'2 from ARG-58 to VAL-72 and finally R'3 from ASP-85 to ASP-95. The aforementioned flexible regions are structurally composed of loops that play a key role in molecular recognition.
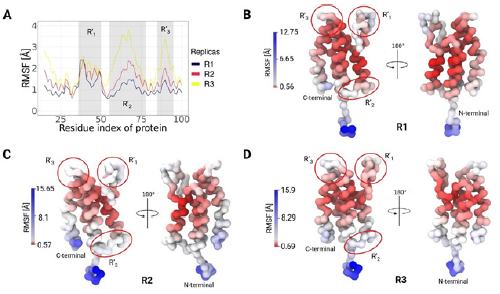
Figure 2 RMSF observed in the bromodomain-bromosporine system by MD. A. The bromodomain RMSF is shown except for the terminal regions, where there are three regions of flexibility (R' R'2 and R'3), consistent with the three replicates performed. B, C and D highlight the regions of highest flexibility in the bromodomain, excluding the terminal regions. All replicates were presented coloured by β factor on the structure, where red indicates the most stable regions and blue the most fluctuating ones.
Bromosporine presents several recognition states
The stability and structural changes of the bromodomain and bro-mosporine were evaluated through root-mean-square deviations (RMSD). Figure 3A and 3B indicated different stability values for both; bromodomain presented high values with an average around 5 Å, and bromosporin presented low values 2 Å. The results also indicated that bromosporine presents more than one conformational change. According to the RMSD density plots we noticed three forms of recognition that bromosporin may have in order to interact favourably with the receptor. The obtained RMSD values of the ligand and complex present a correlation because the conformational changes of the complex, specifically the high plasticity of the pocket, cause freedom of movement of the ligand and could generate several rearrangements during the simulation.
Figures 3D, 3E and 3F using a 2D free energy landscape approach revealed that the ligand scans through three different conformational states had similar results obtained based on the RMSD of the ligand. In addition, based on the RMSD values the 3D structure of the receptor and ligand in the initial structure were obtained, as well as the one showing the maximum RMSD. Both structures were aligned for comparison purposes and to observe the regions showing the highest variability. Figure 3C indicates that the greatest variations in the structure, with respect to the initial one, are in the N-terminal and C-terminal regions.
Figure 3 Evolution of the conformational states of the ligand next to the receptor throughout the simulation. A. Comparison of the conformational change in the backbone of the bromodomain of the three replicates. B. Conformational change of the ligand, where at least three conformational states (Sa, S b and Sc) were determined. C. Structural comparison of the bromodomain at the initial time and at the RMSD max value. D, E and F 2D Free energy landscape (FEL) plot of the ligand to the three replicates respectively. All figures contain highlights of the possible conformational states found in the scan.
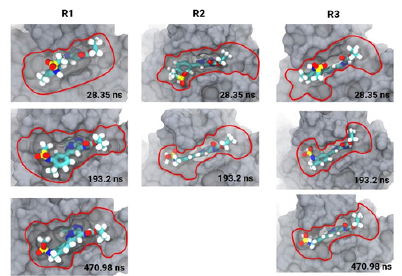
Figure 4 is a set of 3D images showing the bromodomain binding cavity and its associated ligand at three different times. In each image a different conformation of the ligand within the binding cavity is observed. The first image shows the conformation of the ligand at 28 ns with an area of interaction with the receptor determined by the area bounded by the red line. The second image shows a modified conformation of the ligand and the interaction area increases to 193 ns. Finally, the third image shows a new rearrangement of the ligand within the cavity and an increase in the contact area with the receptor. In addition, the images indicate conformational trends of the pocket during the simulation in all replicates, indicating the importance of pocket plasticity.
The relative freedom that can be observed of the ligand during the course of the simulation is due to the constant movement of the loops surrounding the enzyme pocket. The movements allow the ligand to interact with key residues in the complex and start rearranging its atoms until it finds the thermodynamically stable location. The bromosporine at the end of the simulation can fit better because it interacts with the ZA loop which is deeper with in the enzyme pocket than the BC loop [46].
Van der Waals energy key to molecular recognition
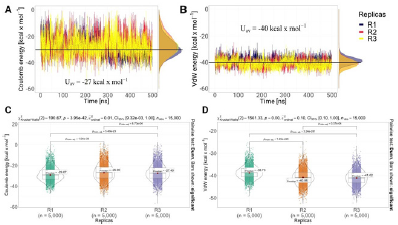
In molecular dynamics simulations, non-bonded energy interactions such as Van der Waals and Coulomb were calculated. Figure 5A and 5B shows the average of the replicas of both energies. Van der Waals interactions contribute more to molecular recognition because they present a strong average energy value of -40 kcal mol-1compa-red to Coulomb interactions with an average weak value of -27 kcal mol-1. Figure 5C shows a Kruskal-Wallis statistical study performed on both interactions to observe the significant differences between each replicate with a value of n=5000. The analyses indicated that there is a significant difference between the replicates in both Van der Waals and Coulomb force.
Free energy
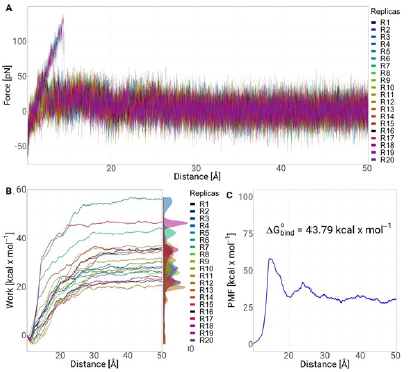
In Figure 6A, we present the average strength profiles as a function of distance obtained from 20 SMD simulations for the ligand bromosporine bound to the bromodomain complex. At the beginning of each simulation (t = 0 ns), the ligand is in a bound state, interacting with the protein residues at the binding site. We observe the separation force of the bromosporine bound to the bromodomain complex. In the 20 replicates performed, it is observed that there is a peak of 175 pN near 5 Å. indicating that there was bond breaking or interactions within the complex. After the peak, near 10 Å of separation of the complex, the force values drop and do not present any additional energy barrier. In addition, the work of the system was studied. Figure 6B showed a trend in all replicates, where at the beginning of the simulation, the work has an exponential trend up to 25 Å. After that, the work continues to increase gradually up to 30 Å and then the system stabilises, causing the work to not increase and generating an asymptote in the replicas. Finally, we calculate the potential of mean force (PMF) using the Jarzynski equation; Figure 6C shows the PMF profiles as a function of stretching distance between the 20 replicates performed along the coordinate set. The profile consists of two peaks, the first one is near 10 Å which is the highest value with 60 kcal mol-1 and the second one starts after 20 Å and reaches a value of 40 kcal mol-1 at a distance of 25 Å. The obtained energy (AG° bind ) value was 43.79 kcal mol-1.
Asparagine and tryptophan key residues in the binding site
Figure 5A shows 3D images of hydrogen bond interactions and hydrophobic interactions with their respective distances. Key amino acids, especially ASN-87 and TRP-93 interacting with bromosporine and surrounding the hydrophobic bromodomain pocket are observed. In addition, Figures 5B, 5C and 5D show the percentage occurrences of hydrophobic, hydrogen bonding and ϖ-stacking interactions respectively of the bromodomain protein amino acids in the three replicates. Hydrophobic interactions indicated that the amino acid TRP-93 is the one that presented the highest frequency, reaching 20 % followed by two Valines at position 36 and 41 that have a similar frequency of 7.5 %. On the other hand, in the hydrogen bond the amino acid ASN-87 was the one that clearly presented the highest and most significant values with respect to the remainder of the amino acids. Finally, ϖ-stacking interactions presented the lowest percentage of occurrence with values less than 1 %. Both of the aforementioned residues provide the major contribution in the binding of the bromosporine inhibitor to the complex. Therefore, these residues may be effective targets for the development of selective bromodomain inhibitors in L. donovani.
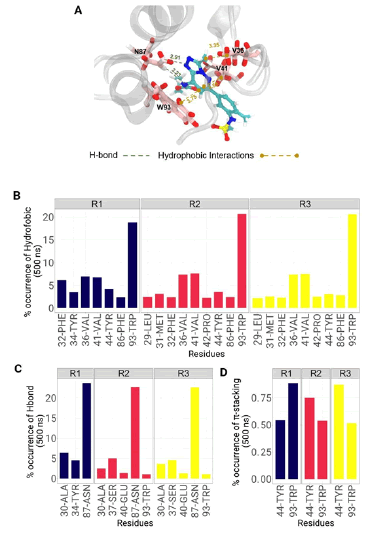
Figure 7 Molecular interactions of the bromosporine-bromodomain system observed during MD. A. The spatial conformation of the system and the key amino acids are shown with the approximate distance in Å, from the crystal structure. B, C, and D show the frequency of occurrence in % of the hydrophobic, H-bond and ϖ-stacking interactions.
Discussion
In this work, we used conventional molecular dynamics and steered molecular dynamics simulations to evaluate the structural dynamics, energetics and recognition mechanism of the bromosporin inhibitor coupled to the bromodomain complex of L. donovani.
The analyses showed that in all replicates there are three regions with high fluctuation values. Regions R'1 and R'3 correspond to the binding loops; BC and ZA. The values are as expected because both belong to the active site that comes in direct contact with the ligand and it has been shown that the movement of the residues is important for ligand recognition in the bromodomain. The results are similar to those obtained by [47] who analysed eight ligand-coupled bromodomain complexes showing high RMSF values at the active site. The R'2 region corresponds in the same way to a loop so it is expected to vibrate more with respect to the helix regions [48].
The RMSD values indicated that the replicates obtained from the bromodomain present values >3.0 Å indicating that they have low stability during the simulation. This may occur due to interaction with the ligand or hydrophobic residues in the loops that may contribute to the instability of the structure [49], [50]. In the case of the ligand results, they showed stability and it could be determined that there are three conformational states. A possible reason for finding multiple states in the ligand is because it is promiscuous and can interact with different structural subfamilies of bromodomains [51]. In fact, it was designed to interact not only with the identified binding site but also with a channel formed by the ZA loop and helix A that is present in almost all bromodomain structures. However, this pocket has rarely been observed to undergo interaction by histone peptide ligands. The observed variations in conformations suggest changes in orientation, position and possible interactions between the ligand and the linker pocket over time. These conformational changes may be essential for the stability and specific interactions between the ligand and the active site. Taken together, the images capture the dynamics of the ligand-cavity complex and provide key information for understanding their interaction at different times in the process [52].
In the case of SMD simulations, the force profiles showed that there is a peak at cleavage of the bromosporin with the bromodomain complex. The observed energy barrier is due to the cleavage of H-bonds, in this case of ASN-87 with the ligand. Subsequently, the values were observed to drop to zero indicating that the ligand completely dissociates from the binding pocket of the receptor [53], [54]. Similar results were observed in the calculations of work required for uncoupling and PMF which after presenting peaks both started to stabilise after 25 Å [55].
Finally, the binding interaction results between bromodomain and bromosporin indicated that in all three replicates there is H-bond formation of the ASN-87 residue [56]. The data are as expected due to previous investigations, where ligands recognise the central cavity of the receptor and anchor via hydrogen bridging to an asparagine residue present in most bromodomains. In addition, it could be determined that in the BC loop there is TRP-93 which together with VAL-41 and VAL-46 located in the ZA loop are key residues in the hydrophobic interactions allowing the ligand to fully enter the binding site [57], [58].
Conclusion
In this work, we performed computer biomolecular simulations of the bromosporin ligand bound to the bromodomain complex. RMSF and RMSD analyses indicated that the bromodomain has greater flexibility than the ligand, causing the ligand to have freedom of movement and producing three conformational states during the simulation. In addition, it was determined that Van Der Waals interactions are key for the recognition of the ligand to the active site to occur. Finally, it could be determined that residues ASN-87 and TRP-93 surround the enzyme pocket and are located in the BC loop of the complex. Therefore, the findings of this study provide useful dynamic information related to conformational alterations and structure-affinity relationship at atomistic levels for new therapeutic strategies towards the bromodomain.

