











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La hemofilia es un trastorno de la coagulación, al cual se le atribuye un origen genético, con un patrón hereditario recesivo ligado al cromosoma X, en donde se encuentran alterados los factores de la coagulación VIII y IX, ocasionando un déficit funcional y cuantitativo que se denomina, respectivamente, hemofilia A y B 1. Estas patologías tienen manifestaciones clínicas muy similares. Además, únicamente la pueden presentar los hombres; las mujeres son portadoras de la enfermedad 1.
Dependiendo de los niveles del factor, se clasifica como severa (< 1 % del valor normal), moderada (1-5 % del valor normal) o leve (> 5 % del valor normal) 2. Se caracteriza por sangrado espontáneo o provocado en articulaciones, músculos u otros tejidos blandos, causando dolor significativo, inflamación y, de no ser tratada, daño permanente 1. El tratamiento de esta condición se basa en la administración del factor de coagulación cuando hay episodios de sangrado (tratamiento a demanda) o de forma regular (tratamiento profiláctico) 2.
Las complicaciones derivadas de esta patología pueden ser incapacitantes y afectar la calidad de vida del paciente de manera significativa, siendo el sangrado articular y la aparición de inhibidores dos de las principales complicaciones que pueden deteriorar gravemente la calidad de vida de los pacientes 3. Por fortuna, se han logrado grandes avances en los últimos años en el manejo de esta condición y aquellos que la padecen pueden llevar una vida relativamente normal 3.
El objetivo es presentar una revisión bibliográfica clara y práctica de la hemofilia, donde se abordan aspectos generales de la fisiopatología, el diagnóstico y el manejo, al igual que las nuevas alternativas terapéuticas en desarrollo para su tratamiento.
Epidemiología
La hemofilia A tiene una incidencia de 1/5000 niños varones nacidos vivos, mientras que para la hemofilia B es de 1/30000. Esta incidencia es casi constante en todas las poblaciones. El tipo A representa el 80 % de los casos de hemofilia, siendo la patología ligada al cromosoma X más frecuente y la segunda en frecuencia de las afecciones hemorrágicas de origen genético, después de la enfermedad de von Willebrand 4-7.
Por otro lado, la prevalencia de hemofilia varía en diferentes regiones, siendo menor en países de ingresos bajos respecto al promedio internacional. Existen múltiples razones para que ocurra esta variación en los reportes a nivel mundial, como son: la falta de capacitación diagnóstica, los pacientes no han sido identificados, falta de acceso a la atención médica, falta de recursos económicos, y poca o nula posibilidad de terapia de reemplazo 8.
En Colombia, según la última encuesta global de la Federación Mundial de Hemofilia (World Federation of Hemophilia Global Survey) de 2016, se reportaron un total de 2059 personas con diagnóstico de esta deficiencia sanguínea (1705 con hemofilia A y 354 casos de hemofilia B), siendo los pacientes entre los 19 y los 44 años el grupo etáreo más frecuentemente afectado (38 % de los casos de hemofilia A y B) 9.
De igual forma, debido a su baja prevalencia, y por ser una condición crónicamente debilitante y amenazante para la vida, el Ministerio de Salud y de la Protección Social clasifica la hemofilia como una enfermedad huérfana e incluso figura dentro de la lista de enfermedades de alto costo para el sistema de salud colombiano 10. Adicionalmente, en el país no se cuenta con rutas o modelos de atención integrales para este tipo de enfermedades; sin embargo, la legislación colombiana le da un enfoque diferencial al abordaje de enfermedades huérfanas como la hemofilia, permitiendo la participación activa en ligas y asociaciones de pacientes, que luchan por la defensa de sus derechos 11.
Patogénesis
En términos generales, existen tres pasos que facilitan la coagulación de un vaso sanguíneo después de una lesión 12:
Vasoconstricción del vaso lesionado.
Activación y agregación plaquetaria para formar coágulo primario.
Coagulación sanguínea.
Para que se dé este último paso deben activarse de manera secuencial una serie de factores, entre ellos el VIII y el IX, que ayudan a la formación de una matriz de fibrina que permite la estabilización del coágulo y favorece la regeneración del vaso lesionado 12.
Como se mencionó anteriormente, la hemofilia es una enfermedad de origen genético de carácter recesivo, ligada al cromosoma X, en donde diferentes mutaciones en los genes que codifican para los factores VIII y IX, para la hemofilia A y B respectivamente, producen un déficit cuantitativo o funcional de los mismos 1. Sin embargo, este patrón hereditario se observa en el 70 % de los pacientes, mientras que el 30 % restante se produce por una mutación de novo; en este último caso, la descendencia del individuo heredará dicha mutación con el mismo patrón recesivo ligado al cromosoma X 1.
Debido a este patrón de herencia, las manifestaciones hemorrágicas suelen darse casi de manera exclusiva en individuos de sexo masculino. Las mujeres también pueden manfiestar la enfermedad en casos excepcionales: fenómenos de inactivación desfavorable del cromosoma X, isodisomía o la concomitancia con un síndrome Turner, entre otras situaciones 5.
Manifestaciones clínicas
La principal manifestación clínica de la hemofilia es la hemorragia, cuyo grado depende del nivel del factor VIII o IX presente en el plasma, usualmente secundaria a traumas en sitios de localización profunda, como articulaciones, músculos y sistema nervioso central (SNC), a diferencia de otras coagulopatías, como la enfermedad de von Willebrand y disfunciones plaquetarias donde el sangrado predomina en mucosas 3,13.
La manifestación hemorrágica más frecuente es la hemartrosis, que afecta principalmente articulaciones grandes como rodilla, codo, tobillo, hombro y cadera; de igual forma, cuando se producen sangrados articulares recurrentes en una misma articulación, se generan cambios atróficos en la misma condición, conocida como “artropatía hemofílica” 5,14. Otras fuentes de sangrado menos comunes incluyen: SNC, sistema gastrointestinal, sistema genitourinario, mucosas nasal y oral (especialmente después de procedimientos dentales) y hematomas en vías aéreas 5.
En cuanto al momento de aparición del primer episodio de sangrado, estudios de cohorte sugieren que cerca del 15 al 33 % de los pacientes lo presentan en el periodo neonatal; sin embargo, los patrones de sangrado que se encuentran en esta población difieren de aquellos que se hallan en individuos de mayor edad 15. En un estudio realizado por R. Kulkarni et al. en 580 neonatos entre los 0 y los 24 meses de edad, se encontró que el 75 % de los infantes fueron diagnosticados en el primer mes de edad, y el 90 %, antes de los 8 meses de edad 15. Asimismo, pacientes con formas más severas de hemofilia pueden manifestarse con sangrado espontáneo, el cual puede ser severo y suele darse en etapas tempranas, incluso desde el nacimiento 16. Por el contrario, casos de hemofilia leve suelen pasar desapercibidos durante largos periodos de tiempo y puede que solo logren identificarse después de que los pacientes son sometidos a procedimientos quirúrgicos 17.
A pesar de que las características clínicas de la hemofilia A y B son muy similares, algunos estudios sugieren que la frecuencia de los episodios hemorrágicos es menor en pacientes con hemofilia B y también sugieren que estos últimos tienen un mejor pronóstico 5,17.
Situación de portadoras
La hemofilia A y B tienen un patrón de herencia recesivo ligado al cromosoma X, afectando casi de manera exclusiva a los hombres; además, las mujeres portadoras tienen un 50 % de riesgo de heredarlo a sus hijos 18. Debido al estado homocigoto de los hombres, estos manifiestan la enfermedad; por el contrario, en las mujeres, por el proceso de lionización aleatoria en uno de los cromosomas X y su condición heterocigota, son principalmente portadoras asintomáticas 19.
La inactivación del cromosoma X en mujeres sintomáticas y mujeres hemofílicas puede verse afectado por algunos mecanismos genéticos que desencadenan efectos clínicos relevantes en estas mujeres portadoras, en comparación con personas sin alteraciones cromosómicas 19.
Algunas mujeres portadoras pueden ser sintomáticas y presentar episodios hemorrágicos en ciertas ocasiones, como: menorragia, hematomas, epistaxis, gingivorragia, entre otros. Se encuentran directamente relacionados con los niveles plasmáticos de factor VIII o IX 20.
La asesoría genética en las familias con casos de hemofilia es importante, para identificar las posibles portadoras antes de tener descendencia, ya que el estudio genético es significativo para confirmar la base molecular de los pacientes, además de ser fundamental en el diagnóstico prenatal 20.
La variabilidad genética en las mutaciones del gen relacionado con la hemofilia ocasiona dificultades en la detección directa del mismo, por lo que se ha desarrollado una estrategia que se basa en la segregación de polimorfismos a nivel familiar, haciendo factible su identificación 7.
Hemofilia adquirida
La hemofilia adquirida o inhibidor adquirido contra el factor VIII, es una enfermedad autoinmune rara, con una incidencia de 1 a 1,5 casos en cada millón de habitantes por año, pero aumenta a 15 casos por millón en mayores de 85 años 21,22.
Esta enfermedad surge de la producción de autoanticuerpos contra los factores endógenos de la coagulación, que lleva al sangrado espontáneo y generalmente severo 23.
Aunque se halla con más frecuencia en hombres mayores de 85 años, puede aparecer en mujeres jóvenes con enfermedades autoinmunes, en el transcurso del embarazo y posparto 21,22. Contrario a la hemofilia congénita, por lo general no manifiesta hemartrosis, y el diagnóstico diferencial incluye coagulación intravascular diseminada crónica, hiperfibrinolisis, púrpura vasculítica o amiloide, entre otras manifestaciones 21.
En 2009 se sugirió que los pacientes con hemofilia adquirida debían ser tratados con corticoesteroides como terapia única, o en combinación con drogas inmunosupresoras, como la ciclofosfamida 22.
Hemofilia infantil
En los menores de 2 años con hemofilia, puede ocurrir una gran cantidad de complicaciones, como sangrados y formación de inhibidores, en respuesta al tratamiento, que tiene impacto a lo largo de la vida. En el estudio realizado por Kulkarni et al. en 580 neonatos, reportaron que el 81 % de los niños experimento algún evento de sangrado durante los primeros dos años de vida, y con relación a la presencia de inhibidores, estos fueron descritos en el 20 % de los niños, de los cuales el 11 % tenía títulos bajos, y el 9 %, altos 15.
En la actualidad, la calidad de vida de los niños con hemofilia ha mejorado gracias a la profilaxis y la posibilidad de realizar el tratamiento en casa; sin embargo, continúan teniendo muchas dificultades, entre ellas la práctica de algunos deportes, lo que genera un impacto sobre su desarrollo sicosocial. Este aspecto puede tener alguna mejoría si se logra capacitar a los pacientes para que tengan autonomía en el manejo de algunas de estas situaciones 24.
Diagnóstico
Para el abordaje diagnóstico del paciente se tienen, como aspectos claves, la elaboración de una historia clínica completa, con énfasis en antecedentes familiares y semiología del sangrado, examen físico adecuado y algunos exámenes de laboratorio confirmatorios. Como pruebas de tamización se cuenta con: hemoleucograma, que permite determinar el tamaño y número de eritrocitos y plaquetas; el tiempo parcial de tromboplastina activado (TPTa); el tiempo de protrombina (TP) y el fibrinógeno 25. Los test de coagulación revelan comúnmente un TPTa prolongado con un TP normal.
En la Tabla 1 se puede ver la comparación de los resultados de laboratorio normal con pacientes con hemofilia 26.
Tabla 1 Resultados de laboratorio normales y en hemofilia
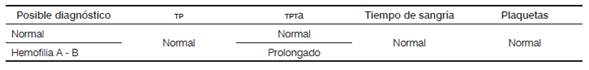
TPTA: tiempo parcial de tromboplastina activado; TP: tiempo de protrombina.
Fuente: adaptada de: Laboratory diagnosis. En: Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, et al.
Guidelines for the management of hemophilia. 2.a ed. Canadá: World Federation of Hemophilia; 2012. p. 31.Con diferentes técnicas de laboratorio se puede realizar la medición de los niveles de actividad plasmática de factores de coagulación. Respecto a la medición de la actividad coagulante (factores VIII o IX), es posible clasificar la patología en tres formas, según la severidad: leve (< 1 % - 1 UI / dL), moderada (1-5 % - 1-5 UI / dL) y severa (5-40 % - 5-40 UI / dL) 26.
Otra parte del diagnóstico incluye el abordaje prenatal. Generalmente, las familias con historia conocida de trastornos en la coagulación acuden a consejería médica y eligen hacer un diagnóstico prenatal en muestras de vellosidades coriónicas o líquido amniótico. Así mismo, se pueden realizar las pruebas posparto, tomando una muestra sanguínea del cordón umbilical. Cuando se tienen valores bajos de factor VIII, es un escenario muy sugestivo de hemofilia A 25,26. El diagnóstico certero se hace por medio de la cuantificación del factor de coagulación deficiente 27.
Tratamiento
Es importante comprender que el objetivo principal sobre el cual se enfoca el tratamiento es evitar el daño articular a largo plazo y mejorar la calidad de vida de los pacientes.
Adicionalmente, el tratamiento está enfocado en prevenir y tratar las hemorragias con el factor de coagulación deficiente. Para esto, en la actualidad se dispone de productos liofilizados, tanto recombinantes como derivados plasmáticos del factor VIII para hemofilia A, los factores IX y VII para hemofilia B, además de complejos activados y adyuvantes, como los antifibrinolíticos 24) (27.
El estándar de oro para el manejo actual de la hemofilia grave se basa en la reposición, de manera profiláctica, del factor faltante, o como tratamiento oportuno ante una situación que lo demande. De acuerdo con la gravedad de la lesión, la dosis calculada del factor VIII se infunde cada 8-12 h, y el factor IX, cada 12-24 h 28.
La profilaxis primaria requiere la infusión de concentrado de factor deficiente entre 2 a 3 veces por semana, iniciando a los dos años de edad, o al presentar el primer sangrado articular; mientras que la profilaxis secundaria se da luego de manifestar compromiso articular.
En la Tabla 2 se enuncian los diferentes tipos de protocolos profilácticos en hemofilia 28.
Tabla 2 Protocolos profilácticos en hemofilia
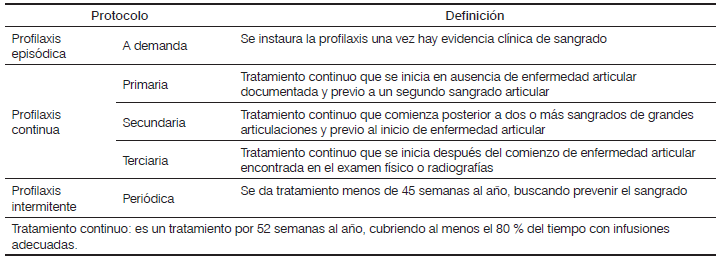
Fuente: adaptada de Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;1-47.
Adaptado de: Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, et al. Guidelines for the management of hemophilia. Haemophilia 2012; 1-47
La dosis que comúnmente se emplea es de 25-40 UI / kg, 2 y 3 veces por semana, para hemofilia B y A, respectivamente (Protocolo Malmo) 28. Sin embargo, existen otras opciones con dosis bajas: 10-20 UI / kg, dos veces por semana, las cuales se recomiendan en países con bajos recursos, con el objetivo de lograr mayor cobertura 29. En Colombia, específicamente, se recomienda la profilaxis para pacientes con diagnóstico de hemofilia severa o moderada con patrón de hemorragia severa 27 (Tabla 3)
Tabla 3 Protocolos de dosis para hemofilia.

Fuente: adaptada de Ministerio de Salud y Protección Social, Instituto de Evaluación Tecnológica en Salud. Protocolo clínico para tratamiento con profilaxis de personas con hemofilia A severa sin inhibidores; 2015; Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: Diagnosis, treatments, and its complications. Lancet 2016;388(10040):187-97, y Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, et al. Guidelines for the management of hemophilia. Haemophilia.2013;19(1)1-47.
A pesar de que existen diferentes protocolos para la profilaxis, es importante individualizar la elección con base a la edad, accesos venosos, tipo de sangrado, actividad y disponibilidad de concentrados del factor. Cuando se empieza a muy temprana edad, una opción es iniciar profilaxis una vez a la semana y escalar paulatinamente, dependiendo de los eventos hemorrágicos y accesos venosos 29.
Iniciar el tratamiento profiláctico primario a edades tempranas se estima una opción óptima, principalmente en población pediátrica con hemofilia severa, ya que trae beneficios, como disminución o cese de episodios de sangrado espontáneo, y prevención de enfermedad articular a futuro; sin embargo, la profilaxis a edades tempranas requiere un acceso venoso central, que permita las infusiones en casa, lo cual puede conducir al riesgo de infección, sepsis y trombosis 25,28,30.
En la hemofilia, además de la profilaxis y la terapia en el momento de los episodios de sangrados, es importante hacer un abordaje integral, que incluye estrategias para el control analgésico. Se ha propuesto una terapia escalonada para este fin 29:
El control efectivo del dolor en esta patología es esencial en la reducción del impacto que ejerce en la calidad de vida, ya que aquel es una realidad en estos pacientes, principalmente en los que tienen hemofilia severa, el cual aumenta la carga de la enfermedad 31.
Otro enfoque necesario para mejorar la calidad de vida es el manejo de la sinovitis crónica en los pacientes que ya han tenido sangrados recidivantes 32. Estos pacientes deben recibir tratamiento específico para esta entidad, antes de seguir con el tratamiento profiláctico, para poder tener mejoría en la funcionalidad articular 32. Inicialmente, se debe identificar la articulación a intervenir, que debe ser aquella que presente malestar clínicamente significativo y que haya tenido mayor que 2 sangrados en los últimos 6 meses 32. Luego, estos pacientes deben comenzar fisioterapia específica para las articulaciones comprometidas 32. El compromiso debe ser evaluado inicialmente por medio de ecografía o resonancia magnética de la articulación, la cual servirá también para el seguimiento en caso de que haya mala respuesta al manejo fisioterapéutico inicial, momento en el que se debe valorar el uso de sinovectomía radioactiva.
Esta terapia es un enfoque alternativo a la cirugía en pacientes con sinovitis con sangrado recurrente, que ha demostrado ser refractario al tratamiento intensivo con concentrados de factor de coagulación 32. Es particularmente una opción terapéutica atractiva para los pacientes con inhibidores o para aquellos que tienen contraindicaciones para la sinovectomía quirúrgica 32.
Otras terapias más convencionales son el uso de embolización angiográfica selectiva o el uso de inyecciones intraarticulares de esteroides, las cuales también deben ser tomadas en consideración cuando se esté tratando a pacientes con síntomas articulares crónicos 32.
Por otra parte, se debe sopesar el tratamiento de la hemofilia adquirida, que consiste en la hemostasis adecuada, erradicación de los autoanticuerpos y tratar la enfermedad subyacente (cuando aplique) 23. Es importante recalcar que el reemplazo con factores exógenos de la coagulación no es efectivo cuando los títulos de anticuerpos son altos, por lo que se deben utilizar a dosis mucho más altas de lo esperado. Adicionalmente, esta terapia debe estar acompañada de corticoesteroides, ya sea solos o en combinación con agentes quimioterapéuticos, como ciclofosfamida o rituximab, para que de esta forma se logren disminuir los títulos de anticuerpos, para hacer más efectiva la terapia de reemplazo 23. En la actualidad se está tomando más a consideración el uso de agentes bypass, que han demostrado ser superiores a la terapia estándar en estos pacientes 23. Sin embargo, aún se requieren más investigaciones que puedan ser extrapoladas a nuestra población con respecto al uso de estas nuevas terapias 23.
Inhibidores
Uno de los mayores desafíos en el tratamiento de la hemofilia es el desarrollo de anticuerpos neutralizantes o inhibidores 33. Estos son aloanticuerpos tipo Inmunoglobulina G (IgG) contra los factores de coagulación VIII y IX, que pueden ser inducidos por la terapia sustitutiva con estos factores, los cuales neutralizan rápidamente la acción de los mismos, lo que hace ineficaz la terapia de reemplazo y expone a los pacientes a complicaciones importantes, aumento en los costos de manejo y en la morbimortalidad 5. Su presencia es más frecuente en individuos con hemofilia A, con una prevalencia del 12 a 35 %, mientras que en la B es de tan solo del 1 al 4 %. De igual modo, se presenta en el 25 al 50 % de los pacientes con hemofilia severa y usualmente se manifiesta en los primeros 50 días de tratamiento 5,33. A pesar de que los mecanismos mediante los cuales se generan estos anticuerpos han sido estudiados ampliamente, aún se desconoce por qué algunos individuos son más propensos que otros a desarrollar esta condición 33. Se ha relacionado la aparición de inhibidores con algunos factores genéticos, como: etnicidad, inmunofenotipo de antígenos leucocitarios humanos (Human Leukocyte Antigen, HLA), defecto molecular en las citoquinas, al igual que con factores no genéticos, como: tipo de producto usado, edad de la primera infusión, vacunaciones, transfusiones previas, entre otros 5.
La inducción de tolerancia inmunológica (ITI) es, hasta el momento, la única alternativa efectiva para restablecer la tolerancia inmunológica al factor, con una tasa de éxito aproximada del 75 %; sin embargo, es una terapia costosa y que implica una infusión diaria de altas dosis de factor, por lo cual es una prioridad actual, en el manejo de la hemofilia (especialmente la hemofilia A), el diseño de estrategias que tengan como propósito evitar la aparición de estos anticuerpos y el manejo de la enfermedad, una vez estos han aparecido. A continuación se describen algunas de ellas 34.
Inducción de tolerancia inmunológica
La ITI hace referencia a la exposición constante a dosis, a menudo altas, de concentrados del factor VIII durante meses o incluso años, con el propósito de inducir tolerancia 33.
Los mecanismos propuestos mediante los cuales se induce la tolerancia incluyen: agotamiento de linfocitos T por sobreestimulación, llevándolas eventualmente a la anergia; inhibición de la diferenciación de linfocitos B específicos contra el factor VIII y la formación de anti-anticuerpos 35.
Eventualmente, tras la terapia exitosa de inducción, los pacientes pueden volver a recibir tratamiento de reemplazo profiláctico y en episodios de sangrado con el factor VIII.
Agentes bypass
Este tipo de agentes permiten la hemostasia al favorecer la formación de trombina sin la necesidad de la participación de los factores VIII o IX 35. Los dos agentes disponibles en la actualidad son el factor VIIa recombinante y el concentrado de complejo de protrombina activado (CCPa). Ambos han mostrado una eficacia hemostática aproximada del 80 %, con pocos eventos tromboembólicos asociados 35.
Estos agentes, por lo tanto, se plantean como una alternativa terapéutica no solo para los episodios agudos de sangrado, sino también para el manejo profiláctico de pacientes con hemofilia y desarrollo de inhibidores mientras son sometidos a terapia ITI, durante esta o después de que la misma ha fallado 34. Sin embargo, la terapia con estos agentes es costosa y su uso simultáneo se asocia a mayores tasas de sangrado, en comparación con la utilización exclusiva de ITI a altas dosis; por lo tanto, el empleo de estos agentes continúa representando un desafío en el tratamiento de pacientes con inhibidores 36.
Nuevos agentes terapéuticos
Debido a las limitaciones tanto de la terapia ITI como en el uso de agentes bypass, en la actualidad se están evaluando nuevos agentes para el manejo de esta complicación. Entre ellos destacan el anticuerpo recombinante humanizado IXa/X (Emicizumab), que imita la actividad del cofactor VIIIa; y el fitusiran, un conjugado de N-acetilgalactosamina, que facilita el transporte de la cadena de ARN interferente ALN-AT3 al hepatocito, la cual interrumpe la producción de ARN mensajero para la síntesis de antitrombina. Esto lleva a menor producción de antitrombina, generándose un fenotipo protrombótico, por el aumento en los niveles de trombina, equilibrándose así el fenotipo hemorrágico existente en la hemofilia 35,37. Pasi et al. encontraron que el uso de fitusiran lleva a disminución de los niveles de antitrombina y aumento en la generación de trombina en pacientes con hemofilia A y B que no han desarrollado aloanticuerpos inhibitorios 38.
De igual forma, en la actualidad se están desarrollando varios ensayos clínicos que evalúan el uso de este medicamento, como el estudio ATLAS (así se denomina la fase 3 del estudio para evaluar la eficacia y la seguridad del fitusiran -ALN-AT3SC- en distintos pacientes con hemofilia A o B, sin anticuerpos inhibidores para factor VIII o IX) y el estudio OLE (Open label extension) 39. Sin embargo, es importante recalcar que, en septiembre de 2017, uno de los participantes del estudio OLE desarrolló trombosis del seno venoso, lo cual llevó a que la casa farmacéutica fabricante del medicamento suspendiera todos los ensayos clínicos que lo incluyeran; no obstante, en noviembre de 2017, el fabricante anunció que había logrado un acuerdo con la Administración de Alimentos y Medicamentos (Food and Drug Administration, FDA) en nuevas medidas de mitigación de riesgos clínicos, incluidas pautas especificadas en el protocolo de los ensayos clínicos, y educación adicional al investigador y el paciente sobre el uso de dosis reducidas de factor de reemplazo o agente de bypass para tratar cualquier hemorragia intercurrente en estudios que incluyeran fitusiran (39.
Por otro lado, al tratarse de una enfermedad ocasionada por el déficit o la disfunción de una sola proteína, la hemofilia es, en teoría, una candidata ideal para el desarrollo de estrategias terapéuticas con terapia génica; sin embargo, el factor VIII es una glicoproteína de gran tamaño, difícil de expresar en niveles terapéuticos efectivos, condición que interfiere con el desarrollo de este tipo de alternativas 34.
Actualmente, el desarrollo farmacéutico está enfocado en la generación de productos de acción prolongada, que permitan disminuir el número de infusiones suministradas a un paciente hemofílico, lo que favorece la adherencia terapéutica y, consigo, la calidad de vida 16. La ingeniería genética es fundamental en la creación de productos mejorados, como es el caso de la glicosilación, que consiste en fusionar proteínas de manera dirigida, con lo que se ha logrado demostrar la mejoría del perfil farmacodinámico y prolongar la vida media de proteínas terapéuticas 16,40.
Por tratarse de una enfermedad monogénica, ha sido estudiada en el desarrollo de la terapia génica, con el objetivo de curar totalmente al individuo, a través de vectores virales (adenovirus). Pese a que es un tratamiento prometedor, en el medio se ha optado por la investigación de presentaciones innovadores de los factores de coagulación, pasando de infusiones a administraciones orales, que mejoran el patrón de adherencia a los medicamentos 25.
Conclusión
La hemofilia, en todos sus subtipos, debe ser considerada una entidad potencialmente mortal, que afecta la calidad de vida de los pacientes y de aquellos que lo rodean. Es un reto diagnóstico que requiere constante indagación y actualización, para que pueda ser tratada de manera efectiva. Se debe fomentar las investigaciones relacionadas con el desarrollo de nuevos productos terapéuticos, como el Emicizumab, o incluso terapias más avanzadas, que se conviertan no solo en una herramienta de tratamiento, sino también en una herramienta curativa, como lo es la terapia génica.
Los médicos tratantes de pacientes con hemofilia deben tener presente uno de los principales desafíos del tratamiento de la hemofilia: el desarrollo de anticuerpos o inhibidores, los cuales neutralizan la acción de los factores de coagulación exógenos, haciendo ineficaz el tratamiento.

