











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkDengue virus (DENV) infection may cause acute systemic diseases. From a number of dengue not-licensed vaccines, the one named CYD-TDV has been recently approved by the World Health Organization (WHO) 1. The CYD-TDV has proven efficacy against confirmed cases in endemic countries of the Americas such as Brazil and México 2 but DENV continues to spread 3,4 widely in Colombia 5. From the 50 to 100 million infection cases estimated worldwide annually 6, roughly 1.5 million occur in Colombia 3.
Dengue can adopt three clinical forms: Self-limited dengue fever (DF), dengue hemorrhagic fever (DHF), and dengue shock syndrome (DSS).
Patients diagnosed with DF present with high fever for two to seven days accompanied by at least two mild hemorrhagic manifestations such as gingival bleeding, petechiae and the like 6, and/or pain (headache, retro-orbital pain, myalgia, or arthralgia, among others). DHF comprises very high fever, hemorrhagic trends, hepatomegaly, or extravasation (thrombocytopenia and/or pleural effusion). Finally, DSS produces a homeostatic alteration characterized by an undetectable pulse before death in DHF/DSS 6-8.
DENV infection has a specific set of mechanisms. Dendritic cells are the first population DENV attaches to 9. DENV binds to the dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC- SIGN) thanks to the E protein on the DENV surface. There is substantial evidence that DC-SIGN is involved with dengue disease development 10,11. Upon DENV infection, pro-inflammatory cytokines that are produced via DC-SIGN may cause apoptosis when patients display cross-reactivity between both platelets and viral antigens 12-15. Some of these cytokines are interleukin-6 and interleukin-8. Interleukin-6 triggers inflammation and activates a wide range of different cells 16. According to Marín, et al., 17 interleukin-6 also induces pro-inflammatory molecules like the interleukin-6- receptor (IL6R) and its soluble isoform (sIL6R). This receptor is expressed by neutrophils and CD4 T-cells 16.
The intercellular mechanisms of DENV infection show patterns associated with pathogens that foster its development and propagation. The intracellular domain of DC-SIGN drives signaling activation of toll-like receptors (TLR), which allow identification of molecular patterns associated with pathogens 9. TLRs activate protection mechanisms and mediate the onset of innate immune effectors18.
The activation of the toll-like receptor 3 (TLR3) inhibits DENV replication in monocytes in vitro resulting in antiviral potential through interferons 18,19.
Although this activation follows direct binding between TLR3 and intermediate double-stranded RNA (dsRNA) from DENV, the conformation of this intermediate dsRNA is essential for DENV replication 7. The latter explains, to some extent, why expression levels of TLR3 among dengue patients are higher than in healthy ones 18.
DC-SIGN is on the nineteenth chromosome in a region of complex alternative splicing sites that generate soluble, membrane-associated, and truncated isoforms 9. Many variants of DC-SIGN have been evaluated for their association with dengue 20 with positive results for some variants within the gene 21. For instance, the SNP rs4804803, allele G, and genotype G/G are highly conspicuous allelic variants found in studies of dengue for the SNP of the rs735239, rs4804803, and rs2287886 located in the DC-SIGN promotor 9. In addition, the haplotype AAG is comprised by the SNPs rs735239-rs4804803-rs2287886 in synteny 9. However, these SNPs are not considered allelic combinations as they are located in the same chromosome.
TLR3 is on the fourth chromosome and encodes the TLR3 protein, a major effector of immune responses to viral pathogens 18,22. TLR3 is found on the surface of the endoplasmic reticulum, endosomes, lysosomes, and endolysosomes 23. The role assessment of TLR3 variants in dengue physiopathology found that the T of SNP rs3775291 is involved not only in disease development but also in its severity 24. Indeed, the frequency of this variant was significantly lower among confirmed DHF cases than healthy controls in India. The polymorphism rs3775291 affects the protein structure and its frequency amounts to 30% between populations with European and Asian ancestry, but not in Africans 25. The 412Phe allele reduced the activation rate in several diseases, such as myocarditis and nasopharyngeal carcinomas, in response to viral infection 22.
The IL6R gene is on the first chromosome and encodes the IL6R/sIL6R isoforms. The sIL6R is produced by alternative splicing at the first exon, right on the rs8192284 variant and by proteolysis of membrane IL6R with the protease ADAM17 26. ADAM17 recognizes the substitution Asp358 A la rs8192284 C, formerly named rs2228145 27. The sIL6R is a multifunctional cytokine essential for the immune response, hematopoiesis, and acute phase reactions. It acts via a heterodimeric receptor of proteins. The expression of interleukin-6 is triggered 100-1,000 times when linked with both IL6R and glycoprotein 130 on the membrane 28. The expression of gp130 is a phenomenon known as interleukin-6 trans-signaling. High levels of sIL6R have been observed in HIV-positive patients vs healthy controls 29.
The genetic variants IL6R, TLR3, and DC-SIGN could actually be used as markers for both susceptibility and prognosis of dengue 20-22,24-27,30. In a context of ancestry mixture, determining the association of these variants with dengue in the Colombian population is non-existent. However, the risk of dengue could be modulated by these variants depending on the ancestry context for specific ethnic groups 31. In this context, our study aimed at establishing the extent to which dengue associated with the genetic variants considered here. To the best of our knowledge, ancestry has not been yet considered in the association between dengue and genetic variants in Colombia. This study evaluated these associations in a sample of Colombian population and the variants in three polymorphisms from three genes of immune sensors.
Materials and methods
Study samples and diagnosis of dengue infection
In an attempt to collect genetically diverse samples, 292 subjects were selected from patients at healthcare institutions from two Colombian departments: Antioquia and Chocó (figure1). As a matter of fact, these subjects guaranteed varying ancestry in these two locations 31,32. All subjects were over 15 years old and recruited during several dengue outbreaks between July, 2009, and December, 2012. Peripheral blood samples (5 ml) were taken after getting the informed consent from all subjects.

The study protocol was approved by the ethics committee at Universidad de Antioquia in June, 2009 (code number 09-12-225). The serum samples were readily centrifuged and kept in frozen storage (-70°C) prior to analysis. The subjects were split into two groups: Controls and cases hinging upon confirmation of dengue as proposed by WHO 6,8. The control group was comprised of 101 subjects without prior records of dengue. Additionally, all subjects in the control group tested negative in the ELISA IgM specific for DENV.
DENV infection in the 191 subjects comprising the case group was confirmed through at least one out of three tests: Enzyme-linked immunosorbent assays ELISA IgM, IgM seroconversion, or reverse transcription polymerase chain reaction (RT-PCR) (table 1). These techniques are detailed by WHO (1997, 2009) 6,8. In short, DENV infection was positively confirmed in 157 samples by the specific IgM antibodies (ELISA Dengue IgM Panbio, Sinnamon Park, Australia) during its acute phase.
Table 1 Diagnosis and demographics of the subjects

1 These blood samples were taken during the acute phase of dengue. Two hundred and ninety two aliquots of blood (one from each participant) were used for DNA extraction, then DNA was destined for variants typing in candidate genes, and ancestry-informative markers.
Sampling was then repeated 15 days later during the convalescent phase, and IgM seroconversion was detected in 30 subjects. Five subjects were not sampled during the convalescent phase.
Finally, four more cases were confirmed by testing DENV infection in 45 subjects following the protocol defined by Lanciotti, et al. (1992) as adapted by Harris, et al. (1998) and modified by Restrepo, et al., (2012) 33-35. The evolution of the symptoms in these latter subjects was of less than five days.
None of the subjects suffered from malaria (thick drop smear). Self-identification served for classifying the subjects as Afro-Colombians or Mestizos.
DNA was isolated from the blood samples following a standard phenol- chloroform protocol 36. Specific genomic regions were amplified through PCR using primers. The ancestry-informative markers (AIMs), its deltas, i.e., allele frequencies reported for ancestral populations, and meticulous typing conditions that were used in this study have been previously detailed 31. The 30 selected AIMs displayed the highest deltas, which in turn were assessed pairwise among the three ancestral populations (European, Amerindian, and African) 37. The distribution of those AIMs throughout the genome is certainly wide.
Most of these AIMs in the battery of polymorphisms of insertion/ deletion and restriction fragment length (RFLP) were resolved using an ABI Prism® 310 Genetic Analyzer (Perkin Elmer-Applied Biosystems). Then, we used two primers per variant in each candidate gene: forward 5’-AGCTTGTCAAATGGCCTGTTG-3’ and reverse 5’-CAGAACAATGGCAATGCAGAG-3’ for IL6R, forward 5’-GGAGCATCAGTCGTTGAAG-3’ and reverse 5’-CTCAACCTAACCAAGAATAA-3’ for TLR3, and forward 5’-AGCCAAAGCTCCTCTAGATC-3’ and reverse 5’-CAGATGGGGTTTCTCCGTGTT-3’ for DC-SIGN. One enzymatic test was performed for resolving one variant in each candidate gene. Specifically, HindIII for rs8192284 A/C”, TaqI for rs3775290 G”/A, and Hpy166II for rs7248637 G”/A (quotes indicate the cut allele). The resulting fragments were then in base pairs: 88, 182; 145, 84; and 138, 88, respectively.
Analysis
First, the data on sex and age were respectively compared through chi-square and Mann-Whitney tests casting case and control groups in 2 x 2 matrixes. Then, three comparative case-control studies were conducted on the 292 subjects using alleles, genotypes, and allelic combinations as independent variables. The proportion of alleles was compared between groups using chi-square after direct counting.
Associations both of genotypes and allelic combinations were estimated by logistic regression using the SNP stat package 38; p-values <0.05 were considered significant and only the lowest were reported. Probability values (p-value), odds ratios (OR), and confidence intervals (CI) were adjusted using the individual African, Amerindian, and European ancestries as co-variables in independent regressions.
Individual ancestry from AIMs data was estimated using the Admixmap program 39. The genotypes were modeled according to inheritance (codominant, dominant, recessive, and overdominant). Genotype deviations from Hardy-Weinberg equilibrium were calculated using SNPstat package. This study focused on developing a model of fixed effects.
Results
Subject diagnosis and demographics
A positive diagnosis for dengue was confirmed in 65.5% (191) of the subjects using the three tests, IgM antibody (157), IgM seroconversion 30, and RT-PCR 4 between the acute and convalescent phase (table 1). RT- PCR was performed for 45 subjects, and four tested positive. The serotypes DENV-1, -2, and -3 were identified within these four subjects and were found to be in complete agreement with those found in official reports from 2008-2009. For the location, the samples were collected according to the procedures established by Restrepo in official reports from 2008-2009 of the Instituto Nacional de Salud (personal communication).
Samples were taken at different times and this could be the reason for the low occurrence of viral infections (9%). For subjects tested withRT-PCR, samples were often collected after more than four days of symptoms evolution during the acute phase of dengue. This is a phase in which viremia starts diminishing, making detection of the viral genome rather complex 40. The male-female ratio was 48.5% and 51.5%, which suggests a non-significant difference. The Mestizo was the most common ethnic group and showed differential ancestry proportions when compared with Afro-Colombians. Mestizos displayed a high proportion of European ancestry (0.679±0.119) with low levels of Amerindian (0.176±0.069) and African (0.154±0.117) ancestries. Among Afro- Colombians, the proportion of African ancestry was relatively high (0.733±0.219), but low for the European (0.178±0.168) and the Amerindian(0.089±0.100).
Allelic variants association with dengue
Every minor allelic frequency was >1% (table 2). Stratification analysis revealed that three allelic variants associated significantly with dengue, two in Afro-Colombians and one in Mestizos (table 3).These associations posed one protective factor and another one for risk of dengue in Afro-Colombians while only one single risk factor for Mestizos. In Afro-Colombians, the C allele of IL6R rs8192284 presented a lower risk, while a higher risk was present in the A allele of DC-SIGN rs7248637. The A allele of TLR3 rs3775290 displayed the most significant association with the risk of dengue for Mestizos. No significant difference was observed between the allelic frequencies in subjects. The data loss rate was minimal.
Table 2 Allelic frequencies and Hardy-Weinberg equilibrium for all subjects and after stratification by self-identification

SNP: Single nucleotide polymorphism
rs: Reference sequence
MAF: Minor allele frequency
* MAF is displayed for each ancestral population as reported by NCBI.
Table 3 Allelic variants association with dengue. Columns display associations for all subjects after stratification by self-identification.
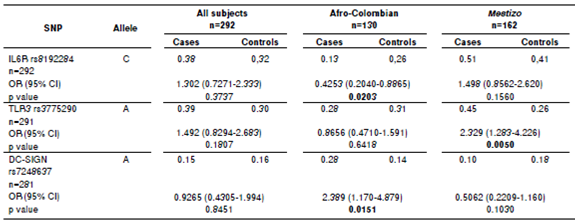
SNP: Single nucleotide polymorphism rs: Reference sequence
OR: Odds ratio in chi-square test
CI: Confidence interval; significant p values are in bold.
Table 4 Association of genotypic variants with dengue for all subjects and after ancestry adjustment

SNP: Single nucleotide polymorphism
rs: Reference sequence
OR: Odds ratio in chi-square test
CI: Confidence interval. Significant p values are in bold.
Table 5 Association of allelic combinations with dengue for all subjects and after ancestry adjustment

* The order of nucleotides in the allelic combinations is based on the order of polymorphisms in the chromosomes rs8192284 - rs3775290 - rs7248637. OR: Odds ratio in chi-square test
CI: Confidence interval; significant p values are in bold
Genotypic variants association with dengue
The risk of dengue for specific ancestry contexts was studied by identifying genotypic variants association with dengue. The three loci remained in Hardy-Weinberg equilibrium in all subjects. The frequency of the genotype A/A in TLR3 was higher in cases than in controls, either with or without ancestry adjustment. The frequency of the genotype C/C in IL6R was also higher in cases than in controls, but only when adjusted by the Amerindian component and with no adjustment (table 4). No significant association of rs7248637 genotypes for cases and controls was found. Eight associations with genotypes were found between cases and controls in all subjects.
Variants of allelic combinations associated with dengue
Two associations between dengue and the allelic combination CAG were observed (table 5). This association was significant for the Amerindian context with and without ancestry adjustment. The CAG combination displayed higher frequency in cases than in controls. No significant association between allelic combinations and dengue was found after adjusting for either European or African ancestries.
Discussion
This study established associations of genetic variants in IL6R, TLR3,and DC-SIGN with dengue after using ancestry or auto-identification control techniques. To the best of our knowledge, this is the first study in America that evaluates the association between dengue and three SNP, namely, rs8192284, rs3775290, and rs7248637, which are located in the encoding genes IL6R, TLR3, and DC-SIGN, respectively. These encoding genes are sensors of the immune system for dengue. The hypothesis evaluated the association of the genetic variants with the susceptibility to suffer dengue in different Colombian ethnic groups or ancestries. To prove this hypothesis, a confirmed dengue group was contrasted using the infection diagnosis in a case-control study of sampled Colombian population (table 1).
We determined African, European, and Amerindian ancestries against Afro-Colombian and Mestizos. The ancestral genetic context differences may influence dengue 41,42. In fact, results of this study concur well with Silva (2010) and Galanter (2012) 32,41 by confirming that specific genetic variants may act as markers for risk susceptibility of getting dengue in different ethnic groups or ancestries as shown in previous studies 10,24.
A total of 191 samples of dengue associated strongly with the genetic variants in SNP rs8192284analyzed for IL6R, SNP rs3775290 for TLR3, and SNP rs7248637 for DC-SIGN (table 3, 4, and 5). Thus, the effect of the three SNPs on the physiopathology should be strong, since this genetic association was found within a small sample. Evidently, the polymorphisms regulate transcription for both receptors and other genes of the immune system that participate in viral recognition and interferon gamma production as a response to DENV infection 19. Additionally, the risk allele combination for dengue CAG found in IL6R/TLR3/DC-SIGN (table 3) was significant without ancestry adjustment and also when controlling for the Amerindian component (p<0.05). A similar trend was found when controlling for both European and African components, although it was not statistically significant. The single most marked observation to emerge from the analysis of allelic combinations was acknowledging the interdependence of all polymorphisms 43.
From the point of view of the analyses for alleles and genotypes, variants associated repeatedly with dengue. A comparison between Afro-Colombian and Mestizo ancestry groups showed that the proportion of the allele C was significantly different (p<0.05) in cases and controls (table 3). The dengue associated with SNP rs8192284 when the genotype C/C was considered before and after controlling for Amerindian ancestry. These repeated associations between variants and dengue could be explained by the likely participation of SNP rs8192284 in the development of joint pain during the acute phase of a number of diseases 6. Additionally, these variants contribute to inflammation amplification among dengue patients via sIL-6/mIL- 6 as reported for SNP rs8192284 in other pathologies 26.
The association between dengue and the variants of SNP rs8192284 for IL6R has not yet been fully established. Notwithstanding, the strong role of the encoding IL6R in the evolution of other diseases like HIV-I as regards viral aetiology has been widely addressed 29. In fact, the evidence on the role of thealleleCinincreasinginflammationforanumberofdiseasesiscompelling. For example, allele C strengthens the development of serious illnesses such as arthritis and myeloma44,45.
The genetic determination of rs8192284 is explained by the enhanced transcription of dendritic cells up to more than 1000 fold for the IL-6. Also, the proteolytic receptor binds to the membrane (mIL-6) just in the alanine 358 that is encoded by the allele C and linked to interleukine-6 28. The association between the variants in genes IL-6, TNFA, INFG and the interleukine-6 levels found in dengue cases has been previously established in Colombia 46. Through linkage disequilibrium, the SNP rs8192284 has also displayed synteny of rs4537545 with other variants in genes IL6/IL6R/pg130in a Caucasian Italian population 47. Naturally, the variants in gene IL-6 weigh the severity of DHF46. On the other hand, the inflammatory signal from dendritic cells is amplifiedbythesolubleformofthereceptor(sIL6-R),whichresultsinremote activation of synoviocytes, chondrocytes, and a number of organs16,48,49.
With regards to SNP rs3775290 for TLR3, the allele A, genotype A/A, and allelic combination CAG were significantly different between cases and controls (p<0.05). As reported by Alagarasu, et al., 24 the evidence we found confirms the contribution of TLR3 polymorphisms to the development of dengue disease for populations in admixture like the Indian. From the point of view of the variability in those allelic variants, it would appear that there are factors for both risk of and protection against developing dengue (table 3). Several authors have challenged this view on the grounds that these factors could change depending on the context of genetic admixture for the ethnic groups to be compared 31,41,42. However, as put forward by Zapata, et al., 12, the variability in alleles, genotypes, and allelic combinations could beexplained by specific differences in the immune response displayed by different ethnic groups. This is consistent with the differences in susceptibility to dengue found between ethnic groups in South America 41. Our results for genotypic distributions and allelic combinations have a number of similarities with these authors (table 3 and 4). An increasing number of studies have recently found evidence that the SNPrs3775290 is strongly associated with virus-related diseases for ethnic groups with a high proportion of either European or African ancestry 50,51.
The role of SNPrs3775290 in viral a etiology has received much attention in the last few years 52. For example, interferon-stimulated response elements were found whilst studying the promoter sequence of TLR3 insilico30.There is evidence to support the hypothesis that this promoter affects the response of TLR3 and other proinflammatory cytokines.Thes ynergy of the proinflammatory cytokines with TNFA and INFG results inenhanced TLRs production30.The TLR3 abundance in endosomes of both dendritic and T cells during dengue development is essential for the immune system to counteract the viral infection 23. In addition, the synonym change F459F encoded by SNP rs3775290 is in strong linkage disequilibrium with rs3775291 among Chinese and Polish population 30,51. In both cases, the disease was triggered by a viral infection (HCMV) with severe implications for its development. Evidence of the linkage between rs3775290 and other functional variants that code no synonym changes in TLR3 for Colombian population is lacking. Further research needs to be carried out to establish whether rs3775290 is in synteny with rs3775291.
As regards DC-SIGN, it plays a major role in sensing the viral infection and mediating pathogenesis control (22). The SNP rs7248637 is important owing to its influence on the DC-SIGN transcription. The SNP rs7248637 lies in the 3’-untranslated region of the DC-SIGN that happens to determine the transcript half-life(53).The development of other viral infections is of ten rapid when transcript levels decrease due to escape to the immune system53. The SNP rs7248637 is also involved in the regulation and recognition of a micro-ARN of interference (miARN) 53, which is capable of modulating the risk against getting a disease in response to viral infections for the African population. As anticipated, our results show that rs7248637 greatly relates to dengue development due to its functional effects.
Worldwide, dengue conspicuously associates with a number of variants for DC-SIGN10,21 because of the aforementioned mechanisms. The association of those variants for DC-SIGN with dengue is more assertively controlled by ancestry than by self-identification 31,42. DC-SIGN supports a variety of primary immune responses. Furthermore, other variants for DC-SIGN could decrease the platelet adhesion that leads to thrombocytopenia 9. DENV infection promotes the destruction of both platelets and megakaryocytes in vitro12. It is thus highly likely that DENV infection impairs homeostasis regulation in vivo.
This study has given an account of the association between dengue and the variants in SNPs rs8192284 for IL6R, rs3775290 for TLR3, and rs7248637 for DC-SIGN. This means that the genetic component that affects the variability in dengue progression is partially comprised by the variants for the genes IL6R, TLR3, and DC-SIGN. Considerable insight has been gained into genetic markers for dengue prediction, for example, therapeutic targets.
Further work could evaluate the role of other variants in the antiviral response associated with IFNs production through TLR34. Zapata, et al., 12 have shown that enhanced production of interferon gamma favors the increase in megakaryocytes that take part in the possible development of dengue coagulopathies. Additional work may look at the association of dengue with all the variants from the polymorphisms in the entire TLRs family, especially with TLRs3/4/7/9. More functional studies are also needed to establish the role of each TLR in the physiopathology and severity of dengue in admixed populations such as the Colombian one.
Our findings may have a number of medical applications. One could be preventing dengue disease in African, European and Amerindian ancestry contexts or ethnic groups by looking at SNP frequencies associated with the risk of getting dengue, which can be assessed through the count of risk alleles in populations or individuals. This knowledge could lead to designing strategies based on non-genetic risk factors such as the use of bed nets and mosquito population control. A second application could be long-term evaluations of associations between the IL6R, TLR3, and DC-SIGN variants and other phenotypes related to DENV infection, such as the progression of hemorrhagic dengue, plasmatic extravasation, and cytokines production by genic expression assays, as well as other functional molecular studies.

