










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cardiología
Print version ISSN 0120-5633
Rev. Col. Cardiol. vol.12 no.3 Bogota Sept. 2005
Efectos pleiotrópicos de las estatinas
Características farmacológicas útiles en la prevención, tratamiento y regresión de la enfermedad cardiovascular
Pleiotropic effect of statins
Useful pharmacological characteristics in prevention, treatment and regression of cardiovascular disease
Darío Echeverri, MD.; Lorena Buitrago, MB., Félix R. Montes, MD.
Laboratorio de Investigación en Función Vascular - Fundación Cardioinfantil - Instituto de Cardiología, Bogotá, DC., Colombia.
Correspondencia: Darío Echeverri Arcila, MD., Calle 163 A No. 28-60, Servicio de Hemodinamia- Laboratorio de Investigación en Función Vascular, Teléfonos: (57-1) 6791192 - (57-1) 6672727 Ext.: 1114 y 4322 - Fax: (57-1) 6717506, Bogotá, DC., Colombia - Correo electrónico: decheverri@cardioinfantil.org, funcionvascular@cardioinfantil.org
Los resultados benéficos de las estatinas en el manejo de la hipercolesterolemia en los múltiples estudios clínicos, han demostrado, además, efectos no relacionados con la acción hipolipemiante. Estudios experimentales in-vitro y ex-vivo han documentado una gran evidencia de efectos tales como incremento en la expresión de óxido nítrico y efectos anti-inflamatorios, inmunomodulatorios, anti-trombóticos, anti-proliferativos y anti-oxidantes los cuales reciben el nombre de pleiotrópicos. Los potentes efectos hipolipemiantes y pleiotrópicos podrían explicar los beneficios en aterosclerosis, hipertensión arterial, diabetes mellitus, estenosis aórtica, psoriasis, esclerosis múltiple y rechazo post-transplante entre otras patologías. Sin embargo, la cantidad de información experimental a favor de estos efectos, debería estimular a la iniciación de mejores estudios para clarificar de una manera mayor el significado clínico.
Palabras clave: ateroesclerosis, hiperlipidemia, inflamacion, efectos pleiotrópicos, estatinas
Among the beneficial results of statins in the treatment of hypercholesterolemia, other effects not related to the hypolipemic action have been demonstrated in multiple studies. Experimental in vitro and ex-vivo studies have documented a great evidence of different effects such as an increment in the nitric oxide (NO) expression, as well as anti-inflammatory, immunomodulatory, anti-thrombotic, anti-proliferative, and anti-oxidant effects. These are the pleiotropic effects. The potent hypolipemic and pleiotropic effects could explain the benefit in atherosclerosis, arterial hypertension, diabetes mellitus, aortic stenosis, psoriasis, multiple sclerosis and post-transplant rejection, among other pathologies. Nevertheless, the amount of experimental information in favour of these effects should stimulate the initiation of better studies in order to clarify in an accurate way its clinical significance.
Key words: atherosclerosis, hyperlipidemia, inflammation, pleiotropic effects, statins.
Introducción
En las últimas décadas, se ha logrado un progreso significativo en el entendimiento de la interrelación entre los desórdenes lipídicos y la prevención de la enfermedad coronaria isquémica, y en este sentido, la identificación de nuevos agentes terapéuticos ha sido el objetivo fundamental de científicos. Los inhibidores de la enzima hepática HMG-CoA reductasa (estatinas), pueden inducir importantes reducciones en los niveles de colesterol plasmático y por ello se consideran como los medicamentos de elección para el tratamiento de la hipercolesterolemia. Importantes estudios han demostrado que las estatinas pueden inducir regresión de la aterosclerosis, así como reducción de la morbimortalidad en pacientes con y sin enfermedad arterial coronaria (1, 2). Los beneficios clínicos de las estatinas usualmente se asumen como resultado de su capacidad de reducir la síntesis de colesterol (3). Estudios realizados por nuestro grupo (93) demostraron, a nivel experimental, que las estatinas pueden inducir regresión de la aterosclerosis temprana y mejoría de la relajación vascular dependiente de endotelio, en un modelo experimental en conejos hipercolesterolémicos.
El mevalonato, producto de la reacción enzimática, es el precursor del colesterol y de compuestos isoprenoides no esteroideos, por lo cual la inhibición de la HMG-CoA reductasa podría resultar en efectos pleiotrópicos (4), diferentes a los hipolipemiantes (Figura). Tales efectos pleiotrópicos incluyen mejoría de la disfunción endotelial, incremento en la biodisponibilidad del óxido nítrico, efectos antioxidantes, propiedades anti-inflamatorias y estabilización de las placas ateroscleróticas entre otros adicionales de interés, que incluyen la capacidad de reclutar células progenitoras endoteliales, una actividad inmunosupresora putativa y una inhibición de la hipertrofia ventricular. Investigaciones indican que algunos de los efectos pleiotrópicos pueden ser no regulados por las propiedades hipolipemiantes de los medicamentos. Otros podrían ser totalmente disociados de la inhibición de la HMG-CoA reductasa y podrían tomar un lugar terapéutico con concentraciones muy bajas.
Esta revisión se focaliza en los efectos pleiotrópicos cardiovasculares de mayor relevancia (Tabla). El entendimiento del amplio espectro terapéutico de las estatinas podría llevar a mejores aplicaciones y al uso adecuado y temprano en síndromes coronarios agudos. Gracias a sus efectos variados, las estatinas se consideran como una de las terapias más importantes en el manejo moderno de la aterosclerosis (5). Ha surgido gran interés en documentar que una terapia farmacológica podría estabilizar la placa aterosclerótica, reducir eventos coronarios agudos y favorecer la regresión de la enfermedad. En medio de este contexto, las estatinas emergen como la mejor terapia sistémica disponible, para el tratamiento y prevención de una enfermedad sistémica como lo es la aterosclerosis (6).
Mejoría de la disfunción endotelial
El trauma endotelial contribuye a la iniciación del proceso aterosclerótico. La disfunción endotelial, como una manifestación temprana del trauma, se asocia con una vasoconstricción paradójica a la acetilcolina dada por el compromiso en la síntesis, liberación y actividad del óxido nítrico. Una respuesta vasomotora anormal dependiente de endotelio, predice la progresión a largo plazo de la aterosclerosis y se asocia con eventos coronarios así como con eventos después de una cirugía vascular (7, 8). De tal manera que no es sorpresa que la disponibilidad de estatinas para mejorar la disfunción endotelial, como efecto de clase, haya recibido mucha atención en los últimos años.
El tratamiento a corto plazo con estatinas ha mostrado mejorar la disfunción endotelial y el incremento de la perfusión miocárdica. En pacientes hipercolesterolémicos con anormalidades en la perfusión miocárdica, el tratamiento con fluvastatina (40 a 80 mg/día) por 6 a 12 semanas, incrementó significativamente la perfusión miocárdica en segmentos isquémicos (5%; p<0,005) (9). En sujetos con colesterol moderadamente elevado (6,2 a 7,5 mmol/L), el tratamiento con 20 mg/día de simvastatina, comparado con placebo, incrementó (p<0,005) la respuesta vasodilatadora a la acetil-colina determinada por el flujo sanguíneo braquial tan temprano como cuatro semanas después del inicio de la terapia (10). Luego de tres meses adicionales de tratamiento, el grupo tratado con simvastatina mejoró (p<0,005) más que lo observado a las cuatro semanas.
En otro estudio (11) se comparó atorvastatina 10 mg/día más una terapia con dieta sola en mujeres hipercolesterolémicas postmenopáusicas. La vasorreactividad de la arteria braquial mejoró significativamente tan temprano como dos semanas luego del inicio del tratamiento en comparación con el grupo control de dieta sola (p<0,001), así como a las cuatro y ocho semanas de seguimiento. Solamente se observó una débil correlación entre los niveles de reducción del colesterol con atorvastatina y la mejoría en la vasorreactividad. En efecto, un pequeño estudio en hombres jóvenes, sanos normocolesterolémicos, indicó mejoría de la función endotelial dentro de veinticuatro horas de tratamiento con 80 mg/día de atorvastatina y reducción rápida con la suspensión del tratamiento después de treinta días (12). El efecto ocurre antes de que los niveles de colesterol sérico y la proteína C reactiva de alta sensibilidad (hsCRP) se reduzcan después de dos días de tratamiento.
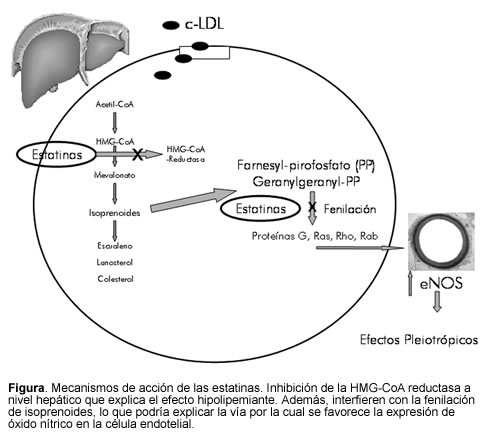
Estos hallazgos dan soporte al concepto por el cual las estatinas ejercen efectos benéficos sobre la función endotelial, que son independientes del grado de reducción del colesterol sérico en el plasma. La terapia con estatinas a largo plazo, también mejora la función endotelial en pacientes con aterosclerosis. Luego de un año de tratamiento, se evaluaron la dieta sola, un régimen para reducción de c-LDL (lovastatina y colestiramina) e hipolipemiantes más antioxidantes (lovastatina y probucol) sobre la vasoconstricción inducida por acetil-colina en arterias epicárdicas (13). En el grupo de estatinas- antioxidantes se observó mejoría en la respuesta vasoconstrictora (p<0,01).
Aún se investiga si la terapia con estatinas tiene un efecto benéfico similar sobre la relajación vascular dependiente de endotelio en la diabetes mellitus. Recientes estudios muestran el impacto de la terapia con estatinas sobre la vasodilatación dependiente de endotelio en la diabetes mellitus tipo 2 (14, 15). Sin embargo, otro estudio con atorvastatina en diabetes mellitus tipo 2, demostró mejoría significativa en la vasodilatación dependiente de endotelio (16). Se informó un hallazgo similar con atorvastatina en pacientes jóvenes con diabetes mellitus tipo 1 y niveles de colesterol normal (17). Los resultados contradictorios de estos estudios podrían justificarse por las diferencias en las dosis del medicamento, el diseño de los estudios, la selección de los pacientes, la medicación concomitante y la tecnología utilizada para la medición de la disfunción endotelial.
Incremento en la biodisponibilidad del óxido nítrico
Las estatinas mejoran la disfunción endotelial en parte por reducir el c-LDL; más específicamente, éstas han mostrado prevenir la «down-regulation» de la enzima óxido nítrico sintasa endotelial (eNOS), la cual cataliza la formación de óxido nítrico a partir de la L-arginina por las LDL nativas (18). La «down-regulation» de eNOS podría ser mediada por la disponibilidad de c-LDL en incrementar la caveolina-1, el principal inhibidor de la actividad de la eNOS (19).
Las estatinas también aumentan directamente la actividad de la eNOS constitutiva y así la biodisponibilidad de óxido nítrico (20). Hay varios mecanismos que pueden estar involucrados, incluyendo una reducción de la caveolina-1 y una mayor abundancia e incremento de Hsp90, que actúa como una chaperona molecular para facilitar la activación a largo plazo de la eNOS (19). Otros mecanismos incluyen la estabilización del ARNm de eNOS (21) y la disminución en la producción de radicales libres que interactúan con el óxido nítrico (22). Las estatinas también interfieren con la fenilación de la Rho-GTPasa por el geranylgeranyl pirofosfato (GGPP), previniendo su traslocación a la membrana celular regulando de manera negativa la actividad de la eNOS (23). La vía de PI3-kinasa/Akt también está involucrada en la regulación del óxido nítrico. Las estatinas fueron capaces de activar la serina/treonina kinasa Akt (proteína kinasa B) en la célula endotelial; de esta manera aumentaron la fosforilación del sustrato Akt para eNOS y produjeron mayor cantidad de óxido nítrico (24).

Efecto sobre la síntesis de endotelina-1
La reactividad vascular es compleja e involucra gran variedad de sustancias y moléculas. El tono vascular depende de un equilibrio entre sustancias vasodilatadoras (óxido nítrico, prostaciclina, etc.) y vasoconstrictores. Uno de los más potentes vasoconstrictores es la endotelina-1 (ET-1), la cual se sintetiza en la célula endotelial y contribuye a desórdenes vasculares agudos, entre ellos a la angina inestable (25), la aterosclerosis avanzada y la disfunción endotelial (26). Las estatinas podrían atenuar la síntesis de ET-1 en forma indirecta al incrementar la disponibilidad de óxido nítrico. Además, se ha descrito que la atorvastatina y la simvastatina reducen la expresión de ARNm de la pre-pro-endotelina-1 y la síntesis de ET-1 (27, 28). La evidencia sugiere que un posible papel de las estatinas en la terapia de las enfermedades cardiovasculares está asociado con el control de los niveles elevados de ET-1.
Efecto antioxidante
El fracaso de los antioxidantes en prevenir la enfermedad coronaria en recientes estudios (29), no invalida la teoría oxidativa de la aterosclerosis. La ausencia de este beneficio podría deberse a dosis inadecuadas, al tiempo de tratamiento o al tipo de antioxidante utilizado (30). Además, el énfasis debe dirigirse a la presencia de eventos agudos y efectos en la interacción entre el estrés oxidativo y la inflamación en el proceso de aterogénesis. En vista del papel central que tienen las LDL oxidadas en la aterogénesis, el efecto antioxidante de las estatinas podría tener un interés mayor.
Además de revertir el efecto inhibitorio de las LDL oxidadas sobre la eNOS, las estatinas también tienen un efecto antioxidante directo sobre las LDL in-vitro y ex-vivo (31, 32). Los metabolitos hidroxy de la atorvastatina, pero no de otros compuestos de la misma familia, inhiben la oxidación de LDL y VLDL así como de las HDL (33). Los metabolitos hidroxy que representan el 70% de la atorvastatina activa en plasma, poseen capacidades lavadoras de radicales libres que pueden contribuir a la inhibición de la oxidación de lipoproteínas. Las estatinas también podrían afectar de modo indirecto los mecanismos oxidativos normales al frenar la capacidad de los macrófagos para oxidar lipoproteínas (34). Asimismo, han mostrado reducir la actividad del receptor CD36 de los macrófagos, reconocido en la oxidación de LDL (35). Los mecanismos de este efecto, se encuentran aún bajo investigación.
Las partículas de LDL oxidadas están cargadas eléctricamente negativas. Posibles causas de la electronegatividad del c-LDL incluyen la glicación y un contenido anormal de ácido siálico (36) y es causa de citotoxicidad. En pacientes con hipercolesterolemia familiar, el tratamiento con 40 mg/día de simvastatina, disminuye significativamente la proporción de c-LDL electronegativo luego de tres meses de seguimiento (29%; p= 0,0002) y pasados seis meses (21%; p<0,0001). Durante seis meses de terapia con simvastatina, la cantidad de colesterol transportado en c-LDL electronegativo continúa en reducción, hasta lograr un 60%. Mucho más temprano, como al mes de tratamiento, se hicieron evidentes cambios importantes en los parámetros de lípidos séricos (36). Estos hallazgos sugieren que la terapia con estatinas a largo plazo en humanos, puede llevar a una reducción progresiva en el potencial aterogénico asociado con el c-LDL electronegativo.
La aterosclerosis se caracteriza por el depósito de macrófagos y la formación de células espumosas en la íntima arterial, que se originan a partir de la diferenciación de monocitos sanguíneos circulantes que han capturado partículas LDL oxidadas. La terapia con estatinas ha sido estudiada en sus efectos sobre la captura de c-LDLox por parte de estas células inflamatorias en la placa aterosclerótica (37). Se demostró que las estatinas suprimen la «up-regulation» de receptores de c-LDL (CD36, SRA-I, SRA-II) en las células. Estos efectos se le atribuyen a las propiedades antioxidantes, ya que se logró reducir la lipoperoxidación lipídica del plasma en 35%, incrementar el estatus antioxidante en 30% y aumentar la actividad de la paraoxanasa sérica en 35%.
Efecto anti-inflamatorio
Gracias al conocimiento obtenido en la pasada década, la importancia de la inflamación en el desarrollo de la aterogénesis ha sido clara. Los niveles elevados de marcadores inflamatorios tales como hs-PCR, interlukina-6, molécula de adhesión intercelular–1 (ICAM-1) y amiloide sérico A (SAA), se han asociado con un incremento en el riesgo para presentar un primer evento o eventos cardiovasculares recurrentes (38, 39). En especial, los niveles de hs-PCR parecen ser el predictor de mayor poder de futuros eventos.
Reducción de PCR sérica
En la actualidad, existe evidencia contundente que afirma que la terapia con estatinas podría atenuar el efecto de la inflamación sobre el riesgo de eventos cardiovasculares. Entre 708 pacientes post-infarto en el Colesterol and Recurrent Events trial (CARE) (38), sujetos con niveles elevados de PCR y SAA (> percentile 90) tuvieron un alto riesgo y se beneficiaron más de la terapia con pravastatina 40 mg/día que aquellos sin niveles elevados de estos marcadores inflamatorios. De base, ambos grupos de sujetos tenían perfiles de lípidos y lipoproteínas en plasma idénticos. La terapia a largo plazo con pravastatina en el estudio CARE también redujo los niveles de PCR en pacientes post-infarto (40). Aunque los niveles medios de base de PCR para el grupo de tratamiento activo y placebo fueron similares, el nivel medio después de cinco años fue 21,6% más bajo en el grupo de pravastatina que en el grupo placebo (p= 0,007).
Los cambios en los niveles de la PCR con el tratamiento de pravastatina no se correlacionaron con la reducción del c-LDL. Estos hallazgos se confirmaron en el estudio con seguimiento a veinticuatro semanas Pravastatin Inflammation/CRP Evaluation trial (PRINCE) (41). Otro estudio comparó la terapia con pravastatina, simvastatina y atorvastatina sobre los niveles de hs-PCR en pacientes con hiperlipidemia combinada (42). Los tres medicamentos, a las dosis utilizadas demostraron tener efectos equivalentes sobre el c-LDL, reducción significativa de los niveles medios de hs-PCR (20% con pravastatina, 23% con simvastatina y 28% con atorvastatina). Estas reducciones no tuvieron correlación con los efectos sobre el c-LDL. Este estudio contrasta con los resultados negativos de una comparación similar que usó diseños paralelos y realizó una exposición durante tres meses en un pequeño número de sujetos en observación (43). En el Atorvastatin versus Simvastatin on Atherosclerosis Progression study (ASAP), la terapia agresiva con atorvastatina (80 mg/día) redujo los niveles de PCR de manera mayor que la terapia convencional con 40 mg de simvastatina (44). Sin embargo, se encontró una correlación significativa en el análisis de univarianza entre la reducción en los niveles de PCR y el grosor medio de la íntima (IMT) en los segmentos de la arteria carótida. Un reciente estudio mostró que la simvastatina redujo los niveles de PCR dentro de los catorce días de tratamiento (45), independientemente de la reducción de los niveles de c-LDL. Las reducciones rápidas en hs-PCR con la terapia de estatinas podrían explicar, en parte, los efectos benéficos tempranos de estos medicamentos en síndromes coronarios agudos (SCA) (46).
Reducción de moléculas de adhesión
Las moléculas de adhesión y quimioatrayentes juegan un papel importante en el proceso de inflamación vascular y aterosclerosis (47). Ellas median la adhesión y migración de leucocitos al espacio subendotelial como parte fundamental del proceso aterogénico. Es posible medir las diferentes moléculas de adhesión en el plasma y estudiar su interacción in-vitro con las integrinas que se encuentran en la superficie celular de las células inflamatorias.
Las estatinas parecen reducir la adhesión celular y las moléculas quimiotácticas e inhibir la actividad de las integrinas. Sin embargo, algunos estudios muestran resultados inconsistentes. En un estudio no controlado, la terapia hipolipemiante agresiva con simvastatina y atorvastatina fue asociada con una reducción en la E-selectina soluble pero no con moléculas de adhesión vascular celular (VCAM) o moléculas de adhesión intercelular (ICAM) solubles (48). Recientemente, se halló que la atorvastatina y la simvastatina reducen significativamente la E-selectina, la P-selectina y el ICAM-1, pero la simvastatina incrementó el VCAM-1 soluble (49). Otra reciente comparación entre estatinas en altas dosis, mostró solamente pequeños e inconsistentes efectos de los medicamentos sobre los niveles de ICAM-1 (50). La terapia con fluvastatina en pacientes con hipercolesterolemia, redujo los niveles circulantes de P-selectina e ICAM-1; este efecto parece ser independiente del efecto hipolipemiante (20). Un reciente estudio, demostró que una estatina modificada sin efecto inhibitorio sobre la HMG-CoA reductasa hepática, podría tener un efecto anti-inflamatorio potente y selectivo (51). Este hallazgo provee conocimientos acerca de que los efectos pleiotrópicos de las estatinas podrían ser totalmente disociados de la inhibición de la síntesis de colesterol.
Estabilización de la placa aterosclerótica
Existen varios mecanismos que podrían tenerse en cuenta para el efecto estabilizador de placa por las estatinas; esto se demostró en importantes estudios en modelos animales (52). La reducción del c-LDL podría contribuir a la reducción del tamaño del núcleo lipídico (53), a la inhibición de la captura de c-LDL oxidado por CD36 (54), a la depuración de receptores de c-LDL oxidado similar a la lectina (55, 56) y a la inhibición de las propiedades oxidativas de los macrófagos (34). Estos efectos de las estatinas podrían, teóricamente, contribuir a reducir la formación de células espumosas.
Los niveles elevados de varios marcadores de la cascada inflamatoria en el plasma, han mostrado tener un valor predictivo de riesgo de ruptura de placa aterosclerótica. Estos marcadores incluyen P-selectina, interleukina-6, factor de necrosis tumoral a, ICAM-1 soluble y hs-PCR (47). El efecto benéfico de las estatinas sobre el proceso inflamatorio ha sido discutido previamente. El debilitamiento de la cápsula fibrosa en placas inestables o placas vulnerables, se asocia con un incremento en la producción de metaloproteinasas de matriz (MMP) por parte de los macrófagos. En cultivo de macrófagos, la fluvastatina redujo la actividad de MMP-9 entre 20% y 40% (57). En estudios en humanos (58), la pravastatina cambió la composición de las placas en arterias carótidas de una manera que favoreció su estabilización. Pacientes con estenosis de arterias carótidas, recibieron pravastatina 40 mg/día o ninguna terapia por tres meses antes de una endarterectomía carotídea. Las placas removidas del grupo tratado con la estatina, fueron compuestas de una menor cantidad de contenido lipídico y c-LDL, y menor cantidad de macrófagos y células T. Estas placas tenían un alto contenido de colágeno y demostraron menor inmunorreactividad de MMP-2 que las placas del grupo control. Además, la apoptosis se redujo de manera significativa y la inmunorreactividad al inhibidor tisular de metaloproteinasa –1 (inhibidor potente de MMP-1 y MMP-9), incrementó en forma significativa en el grupo con pravastatina en comparación con el grupo control (58).
Efectos adicionales
Efectos anti-trombóticos
Existe una aceptable evidencia en la cual se demuestran propiedades anti-trombóticas de las estatinas, que en la mayoría de los casos no se encuentran asociados con los cambios en el perfil lipídico (59). Estos efectos, comprenden la reducción en la expresión del factor tisular (TF), la disminución en la generación de trombina y la atenuación de varios factores pro-coagulantes catalizados por la trombina, tales como fibrinógeno, activación del factor V y el factor XIII, que incrementan la expresión de trombomodulina, y están principalmente atribuidos a la inhibición de la isoprenilación de proteínas.
En resumen, las estatinas han demostrado reducción en la expresión de TF (60-62), en la producción y activación de FVII (63, 64) y en la generación de trombina (65-67). Los resultados sugieren reducción de la activación FV, formación de fibras de fibrinógeno y activación del FXVIII (68). Algunos resultados en relación con el fibrinógeno son contradictorios. Para unos investigadores las estatinas parecen no tener cambios en la síntesis de fibrinógeno (69, 70) e incrementar la expresión de trombomodulina (71) y la inactivación del FVa (68). Rauch y colaboradores (72), demostraron que la terapia con estatinas se asocia con reducción de la trombogenicidad, sin afectar los niveles de fibrinógeno, L-selectina, P-selectina y sICAM-1.
Estimulación del reclutamiento de los progenitores de células endoteliales
Las EPC juegan un importante papel en la reparación del trauma celular isquémico (73). Resultados de estudios in-vivo e in-vitro indican que las estatinas actúan, en parte, de manera efectiva sobre el factor de crecimiento endotelial, como citokina fundamental en la regulación de la neovascularización, favoreciendo la diferenciación de las EPC (48). La evidencia sugiere que las estatinas aumentan el nivel de EPC circulantes y promueven su movilización hacia áreas isquémicas (74).
En pacientes con enfermedad coronaria estable documentada, el tratamiento con atorvastatina 40 mg/día por cuatro semanas se asoció con un 1,5 veces de incremento de EPC circulantes en la primera semana de tratamiento, y con un incremento de 3 veces sobre la cuarta semana (74). El tratamiento con atorvastatina estimuló la diferenciación de EPC más que aumentar el número total de células madre hematopoyéticas circulantes. Además, la atorvastatina aumenta de manera significativa la migración de EPC en respuesta al factor de crecimiento vascular endotelial (VEGF).
El significado práctico de estas observaciones, podría aclararse en años futuros, pero sugiere la capacidad de acción de las estatinas en procesos de reparación tisular a través de células madre (75) en tejidos vascular y miocárdico isquémico.
Inmunomodulación
Los mecanismos inmunes han sido identificados como importantes en el proceso de aterogénesis. Un incremento cada vez mayor de evidencia sugiere que las estatinas podrían actuar como inmunomoduladores y que su uso podría tener aplicabilidad en el transplante de órganos y otras condiciones que requieran inmunosupresión.
Mach y colaboradores (76, 77), descubrieron un novedoso efecto de las estatinas como represor efectivo de la expresión del antígeno mayor de histocompatibilidad clase II (MHC-II) dependiente de linfocitos T y proveen una explicación detallada molecular del mecanismo de acción. Sugieren que las estatinas podrían tener un efecto inmunosupresor. Este efecto ha permitido que las estatinas se recomienden luego del transplante de órganos y en el tratamiento de otro tipo de enfermedades donde exista una expresión «aberrante» de MCH-II tales como diabetes mellitus, esclerosis múltiple, artritis reumatoidea, psoriasis y enfermedades inflamatorias crónicas como la aterosclerosis.
Se ha demostrado que el tratamiento con pravastatina, agregado a medicamentos comunes anti-rechazo (ciclosporina, prednisona y azatioprina) después de transplante cardiaco, reduce en forma significativa la frecuencia de rechazos (3 versus 14; p<0,005) e incrementa la sobrevida a los 12 meses, comparado con el grupo control (94% versus 78%; p= 0,025) (78). En transplantes cardiacos, el tratamiento con simvastatina en combinación con medicamentos anti-rechazo y una dieta hipolipemiante, incrementó la sobrevida y redujo la incidencia de enfermedad vascular post-transplante, comparado con dieta sola sobre un periodo de cuatro años (79).
También se ha tenido en cuenta la posibilidad de una actividad inmunosupresora intrínseca de las estatinas. Se encontró que la atorvastatina, la lovastatina (10 mmol/L) y la pravastatina (20 mmol/L) tienen la capacidad de reducir el interferón g e inducir una mayor expresión del MCH-II en células endoteliales y macrófagos humanos (80). Este efecto fue revertido por el mevalonato y fue atribuido al efecto inhibitorio de las estatinas sobre el promotor IV del factor transactivador del MCH-II, llevando a una supresión de la activación de linfocitos T. Este hallazgo podría explicar, en cierto modo, la mejoría en la sobrevida observada con las estatinas en pacientes sometidos a transplante cardiaco, si se logra en el proceso una supresión de las respuestas inmunes de las células T helper-1 (81).
Inhibición de la hipertrofia miocárdica
La hipertrofia ventricular izquierda es un factor de riesgo para la enfermedad arterial coronaria y la insuficiencia cardiaca. La hipertrofia de cardiomiocitos de rata inducida in-vitro por angiotensina II (A-II) fue abolida por la simvastatina. La hipertrofia cardiaca in-vivo, inducida en ratas con infusión de A-II por constricción transaórtica también fue inhibida con el uso de simvastatina (2 mg/kg por 4 semanas) (82). Estos hallazgos corroboran que las estatinas ejercen un efecto protector sobre los órganos, incluyendo riñón (83), páncreas (84, 85), pared vascular y corazón.
Efectos sobre la estenosis aórtica
Los beneficios de las estatinas no se han limitado a manifestaciones de enfermedad macrovascular (enfermedad coronaria, cerebrovascular y vascular periférica). Se ha venido acumulando alguna evidencia en beneficios en enfermedades no vasculares, mediante acciones no-hipolipemiantes, tales como estenosis aórtica, enfermedad de Alzheimer, osteoporosis, prevención de diabetes mellitus, retinopatía diabética y degeneración macular acelerada.
Existen una serie de similitudes entre la aterosclerosis y la estenosis aórtica degenerativa, entre ellas la edad de aparición, el trauma endotelial, los procesos inflamatorios, los depósitos de c-LDL, la angiogénesis y los depósitos de calcio (86-88). Con base en lo anterior, se demuestran efectos benéficos de las estatinas sobre la estenosis aórtica degenerativa. Estos efectos son: reducción del contenido de lípidos en la válvula, reducción de la respuesta inflamatoria, incremento local de la expresión de óxido nítrico y reducción de la apoptosis, de MMP-2, de la respuesta angiogénica y de la expresión de VEGF (89-92). Con base en estos resultados, se defiende el uso de estatinas en pacientes con enfermedad degenerativa de la válvula aórtica con gradientes leves o moderados.
Conclusiones
De acuerdo con el crecimiento en la evidencia, no puede negarse que las estatinas son más que simples medicamentos hipolipemiantes. De otra parte, la posibilidad de que las estatinas tengan efectos pleiotrópicos genera grandes escepticismos en salud; sin embargo, no debe desecharse la vasta cantidad de conocimiento generado en este aspecto.
Algunos de los efectos pleiotrópicos operan independientemente de la reducción del c-LDL, se correlacionan pobremente o no tienen ninguna relación con los cambios en el c-LDL, se obtienen rápidamente y también son rápidamente reversibles con la suspensión del medicamento. Los efectos directos en ausencia de modificaciones del colesterol total o c-LDL se han documentado in-vivo e in-vitro. Los efectos pleiotrópicos de las estatinas y otros fármacos están bajo investigación continua para definir de forma contundente su papel en la prevención de los eventos cardiovasculares. Sin embargo, la gran cantidad de información experimental a favor de estos efectos debería estimular la iniciación de mejores estudios para clarificar de una manera mayor el significado clínico.
Bibliografía
1. Maron DJ, Fazio S, Linton MF. Current perspectives on statins. Circulation 2000; 101: 207-13. [ Links ]
2 LaRosa JC. Statins and risk of coronary heart disease. J Am Med Assoc 2000; 283: 2935-2936. [ Links ]
3. Gotto AM Jr, Grundy SM. Lowering LDL cholesterol: questions from recent meta-analyses and subset analyses of clinical trial data issues from the Interdisciplinary Council on Reducing the Risk for Coronary Heart Disease, ninth Council meeting. Circulation 1999; 99: E1-7. [ Links ]
4. Bellosta S, Ferri N, Bernini F, Paoletti R, Corsini A. Non-lipid related effects of statins. Ann Med 2000; 32: 164-176. [ Links ]
5. Erkelens DW. Modern aspects of atherosclerosis and treatment of lipid disorders. Atherosclerosis 2002; 3 (suppl): 1-2. [ Links ]
6. Ambrose JA, Martinez EE. A new paradigm for plaque stabilization. Circulation 2002; 105: 2000-2004. [ Links ]
7. Schächinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 2000; 101: 1899-1906. [ Links ]
8. Gokce N, Keaney JF Jr, Hunter LM, et al. Risk stratification for postoperative cardiovascular events via noninvasive assessment of endothelial function: a prospective study. Circulation 2002; 105: 1567-1572. [ Links ]
9. Eichstadt HW, Eskotter H, Hoffman I, et al. Improvement of myocardial perfusion by short-term fluvastatin therapy in coronary artery disease. Am J Cardiol 1995; 76: 122A-125A. [ Links ]
10. O’Driscoll G, Green D, Taylor RR. Simvastatin, an HMG-coenzyme A reductase inhibitor, improves endothelial function within 1 month. Circulation 1997; 95: 1126-1131. [ Links ]
11. Marchesi S, Lupattelli G, Siepi D, et al. Short-term atorvastatin treatment improves endothelial function in hypercholesterolemic women. J Cardiovasc Pharmacol 2000; 36: 617-621. [ Links ]
12. Laufs U, Wassmann S, Hilgers S, et al. Rapid effects on vascular function after initiation and withdrawal of atorvastatin in healthy, noncholesterolemic men. Am J Cardiol 2001; 88: 1306-1307. [ Links ]
13. Anderson TJ, Meredith IT, Yeung AC, et al. The effect of cholesterol lowering and antioxidant therapy on endothelium-dependent coronary vasomotion. N Engl J Med 1995; 332: 488-493. [ Links ]
14. van de Ree MA, Huisman MV, de Man FH, et al. Impaired endothelium-dependent vasodilatation in type 2 diabetes mellitus and the lack of effect of simvastatin. Cardiovasc Res 2001; 52: 299-305. [ Links ]
15. van Etten RW, de Koning EJ, Honing ML, et al. Intensive lipid lowering by statin therapy does not improve vasoreactivity in patients with type 2 diabetes. Arterioscler Thromb Vasc Biol 2002; 22: 799-804. [ Links ]
16. Tan KCB, Chow WS, Tam VHG, et al. Atorvastatin lowers C-reactive protein and improves endothelium-dependent vasodilatation in type 2 diabetes mellitus. J Clin Endocrinol Metab 2002; 87: 563-568. [ Links ]
17. Mullen MJ, Wright D, Donald AE, et al. Atorvastatin but not L-arginine improves endothelial function in type 1 diabetes mellitus: a double-blind study. J Am Coll Cardiol 2000; 36: 410-416. [ Links ]
18. Martínez-González J, Raposo B, Rodríguez C, et al. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibition prevents endothelial nitric oxide synthase down regulation by atherogenic levels of native LDLs: balance between transcriptional and posttranscriptional regulation. Arterioscler Thromb Vasc Biol 2001; 21: 804-809. [ Links ]
19. Feron O, Dessy C, Desager JP, et al. Hydroxy-methylglutaryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation 2001; 103: 113-118. [ Links ]
20. Romano M, Mezzetti A, Marulli C, et al. Fluvastatin reduces soluble P-selectin and ICAM-1 levels in hypercholesterolemic patients: role of nitric oxide. J Invest Med 2000; 48: 183-189. [ Links ]
21. Laufs U, La Fata V, Plutzky J, et al. Up regulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation 1998; 97: 1129-1135. [ Links ]
22. Wassmann S, Laufs U, Ba¨umer AT, et al. HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension 2001; 37: 1450-1457. [ Links ]
23. Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem 1998; 273: 24266-24271. [ Links ]
24. Fuhrman B, Koren L, Volkova N, et al. Atorvastatin therapy in hypercholesterolemic patients suppresses cellular uptake of oxidized-LDL by differentiating monocytes. Atherosclerosis 2002; 164: 179-185. [ Links ]
25. Kureishi Y, Luo ZY, Shiojima I, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nature Med 2000; 6: 1004-1010. [ Links ]
26. Zeiher AM, Goebel H, Scha¨chinger V et al. Tissue endothelin-1 immunoreactivity in the active coronary atherosclerotic plaque: a clue to the mechanism of increased vasoreactivity of the culprit lesion in unstable angina. Circulation 1995; 91: 941-7. [ Links ]
27. Lerman A, Edwards BS, Hallett JW et al. Circulating and tissue endothelin immunoreactivity in advanced atherosclerosis. N Engl J Med 1991; 325: 997-1001. [ Links ]
28. Lefer AM, Campbell B, Shin YK et al. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation 1999; 100: 178-184. [ Links ]
29. Lefer DJ, Scalia R, Jones SP et al. HMG-CoA reductase inhibition protects the diabetic myocardium from ischemia reperfusion injury. FASEB J 2001; 15: 1454-1456. [ Links ]
30. Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20 536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360: 23-33. [ Links ]
31. Witztum JL, Steinberg D. The oxidative modification hypothesis of atherosclerosis: does it hold for humans? Trends Cardiovasc Med 2001; 11: 93-102. [ Links ]
32. Suzumura K, Yasuhara M, Tanaka K, et al. Protective effect of fluvastatin sodium (XU-62–320), a 3-hydroxy- 3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor, on oxidative modification of human low-density lipoprotein in vitro. Biochem Pharmacol 1999; 57: 697-703. [ Links ]
33. Aviram M, Hussein O, Rosenblat M, et al. Interactions of platelets, macrophages, and lipoproteins in hypercholesterolemia: antiatherogenic effects of HMG-CoA reductase inhibitor therapy. J Cardiovasc Pharmacol 1998; 31: 39-45. [ Links ]
34. Aviram M, Rosenblat M, Bisgaier CL, et al. Atorvastatin and gemfibrozil metabolites, but not the parent drugs, are potent antioxidants against lipoprotein oxidation. Atherosclerosis 1998; 138: 271-280. [ Links ]
35. Giroux LM, Davignon J, Naruszewicz M. Simvastatin inhibits the oxidation of low-density lipoproteins by activated human monocyte-derived macrophages. Biochim Biophys Acta 1993; 1165: 335-338. [ Links ]
36. Fuhrman B, Koren L, Volkova N, et al. Atorvastatin therapy in hypercholesterolemic patients suppresses cellular uptake of oxidized-LDL by differentiating monocytes. Atherosclerosis 2002; 164: 179-185. [ Links ]
37. Sánchez-Quesada JL, Otal-Entraigas C, Franco M, et al. Effect of simvastatina treatment on the electronegative low-density lipoprotein present in patients with heterozygous familial hypercholesterolemia. Am J Cardiol 1999; 84: 655-659. [ Links ]
38. Ridker PM, Rifai N, Pfeffer MA, et al, for the Cholesterol and Recurrent Events (CARE) Investigators. Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels. Circulation 1998; 98: 839-844. [ Links ]
39. Lindahl B, Toss H, Siegbahn A, et al. Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease. N Engl J Med 2000; 343: 1139-1147. [ Links ]
40. Ridker PM, Rifai N, Pfeffer MA, et al, for the Cholesterol and Recurrent Events (CARE) Investigators. Long-term effects of pravastatin on plasma concentration of C-reactive protein. Circulation 1999; 100: 230-235. [ Links ]
41. Albert MA, Danielson E, Rifai N, et al. Effect of statin therapy on C-reactive protein levels—The Pravastatin Inflammation/CRP Evaluation (PRINCE): a randomized trial and cohort study. JAMA 2001; 286: 64-70. [ Links ]
42. Jialal I, Stein D, Balis D, et al. Effect of hydroxymethyl glutaryl coenzyme A reductase inhibitor therapy on high sensitive C-reactive protein levels. Circulation 2001; 103: 1933-1935. [ Links ]
43. Joukhadar C, Klein N, Prinz M, et al. Similar effects of atorvastatin, simvastatin and pravastatin on thrombogenic and inflammatory parameters in patients with hypercholesterolemia. Thromb Haemost 2001; 85: 47-51. [ Links ]
44. van Wissen S, Trip MD, Smilde TJ, et al. Differential hs-CRP reduction in patients with familial hypercholesterolemia treated with aggressive or conventional statin therapy. Atherosclerosis 2002; 165: 361-366. [ Links ]
45. Plenge JK, Hernandez TL, Weil KM, et al. Simvastatin lowers C-reactive protein within 14 days: an effect independent of low-density lipoprotein cholesterol reduction. Circulation 2002; 106: 1447-1452. [ Links ]
46. Olsson AG, Schwartz GG. Early initiation of treatment with statins in acute coronary syndromes. Ann Med 2002; 34: 37-41. [ Links ]
47. Blake GJ, Ridker PM. Novel clinical markers of vascular wall inflammation. Circ Res 2001; 89: 763-771. [ Links ]
48. Hackman A, Abe Y, Insull W Jr, et al. Levels of soluble cell adhesion molecules in patients with dyslipidemia. Circulation 1996; 93: 1334-1338. [ Links ]
49. Seljeflot I, Tonstad S, Hjermann I, et al. Reduced expression of endothelial cell markers after 1 year treatment with simvastatin and atorvastatina in patients with coronary heart disease. Atherosclerosis 2002; 162: 179-185. [ Links ]
50. Wiklund O, Mattsson-Hulte´n L, Hurt-Camejo E, et al. Effects of simvastatin and atorvastatin on inflammation markers in plasma. J Intern Med 2002; 251: 338-347. [ Links ]
51. Weitz-Schmidt G, Welzenbach K, Brinkmann V, et al. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nature Med 2001; 7: 687-692. [ Links ]
52. Fukumoto Y, Libby P, Rabkin E, et al. Statins alter smooth muscle cell accumulation and collagen content in established atheroma of Watanabe heritable hyperlipidemic rabbits. Circulation 2001; 103: 993-999. [ Links ]
53. Llorente-Cortés V, Martínez-González J, Badimon L. Esterified cholesterol accumulation induced by aggregated LDL uptake in human vascular smooth muscle cells is reduced by HMG-CoA reductase inhibitors. Arterioscler Thromb Vasc Biol 1998; 18: 738-746. [ Links ]
54. Pietsch A, Erl W, Lorenz RL. Lovastatin reduces expression of the combined adhesion and scavenger receptor CD36 in human monocytic cells. Biochem Pharmacol 1996; 52: 433-439. [ Links ]
55. Umetani N, Kanayama Y, Okamura M, et al. Lovastatin inhibits gene expression of type-I scavenger receptor in THP-1 human macrophages. Biochim Biophys Acta 1996; 1303: 199-206. [ Links ]
56. Li DY, Chen HJ, Mehta JL. Statins inhibit oxidized-LDL-mediated LOX-1 expression, uptake of oxidized-LDL and reduction in PKB phosphorylation. Cardiovasc Res 2001; 52: 130-135. [ Links ]
57. Bellosta S, vía D, Canavesi M, et al. HMG-CoA reductase inhibitors reduce MMP-9 secretion by macrophages. Arterioscler Thromb Vasc Biol 1998; 18: 1671-1678. [ Links ]
58. Crisby M, Nordin-Fredriksson G, Shah PK, et al. Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases, and cell death in human carotid plaques: implications for plaque stabilization. Circulation 2001; 103: 926-933. [ Links ]
59. Undas A, Brummel-Ziedins KE, Mann KG. Statins and blood coagulation. Arterioscler Thromb Vasc Biol 2005; 25: 287-294. [ Links ]
60. Colli S, Eligini S, Lalli M, Camera M, Paoletti R, Tremoli E. VA statins inhibit tissue factor in cultured human macrophages. A novel mechanism of protection against atherothrombosis. Arterioscler Thromb Vasc Biol1997; 17: 265-272. [ Links ]
61. Hilgendorff A, Muth H, Parviz B, et al. Statins differ in their ability to block NF-kappaB activation in human blood monocytes. Int J Clin Pharmacol Ther 2003; 41: 397- 401. [ Links ]
62. Ferro D, Basili S, Alessandri C, Cara D, Violi F. Inhibition of tissue factor-mediated thrombin generation by simvastatin. Atherosclerosis 2000; 149: 111-116. [ Links ]
63. Morishita E, Minami S, Ishino C, et al. Atorvastatin reduces plasma levels of factor VII activity and factor VII antigen in patients with hyperlipidemia. J Atheroscler Thromb 2002; 9: 72-77. [ Links ]
64. Porreca E, Di Febbo C, Amore C, et al. Effect of lipid-lowering treatment on factor VII profile in hyperlipidemic patients. Thromb Haemost 2000; 84: 789-793. [ Links ]
65. Dangas G, Smith DA, Unger AH, et al. Pravastatin: an antithrombotic effect independent of the cholesterol-lowering effect. Thromb Haemost 2000; 83: 688-692. [ Links ]
66. Lacoste L, Lam JYT, Hung J, et al. Hyperlipidemia and coronary disease. Correction of the increased thrombogenic potential with cholesterol reduction. Circulation 1995; 92: 3172-3177. [ Links ]
67. Aoki I, Aoki N, Kawano K, et al. Platelet dependent thrombin generation in patients with hyperlipidemia. J Am Coll Cardiol 1997; 30: 91-96. [ Links ]
68. Undas A, Brummel KE, Musial J, Mann KG, Szczeklik A. Simvastatin depresses blood clotting by inhibiting activation of prothrombin, factor V, and factor XIII and by enhancing factor Va inactivation. Circulation 2001; 103: 2248 -2253. [ Links ]
69. Beigel Y, Fuchs J, Snir M, Green P, Lurie Y, Djaldetti M. Lovastatin therapy in hypercholesterolemia: effects on fibrinogen, hemorrheologic parameters, platelet activity, and red blood cell morphology. J Clin Pharmacol 1991; 31: 512-517. [ Links ]
70. Bickel C, Rupprecht HJ, Blankenberg S, et al. AtheroGene Investigators. Relation of markers of inflammation (C-reactive protein, fibrinogen, von Willebrand factor, leucocyte count) and statin therapy to long-term mortality in patients with angiographically proven coronary artery disease. Am J Cardiol 2002; 89: 901-908. [ Links ]
71. Masamura K, Oida K, Kanehara H, Suzuki J, Horie S, Ishii H, Miyamori I. Pitavastatin-induced thrombomodulin expression by endothelial cells acts via inhibition small G proteins of the Rho family. Arterioscler Thromb Vasc Biol 2003; 23: 512-517. [ Links ]
72. Rauch U, Osende JI, Chesebro JH, et al. Statins and cardiovascular diseases: the multiple effects of lipid-lowering therapy by statins. Atherosclerosis 2000; 153: 181-189. [ Links ]
73. Dimmeler S, Aicher A, Vasa M, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest 2001; 108: 391-397. [ Links ]
74. Vasa M, Fichtlscherer S, Adler K, et al. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation 2001; 103: 2885-2890. [ Links ]
75. Orlic D, Kajstura J, Chimenti S, et al. Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc Natl Acad Sci USA 2001; 98: 10344-10349. [ Links ]
76. Mach F. Immunosuppressive effects of statins. Atherosclerosis 2002 (suppl 3): 17-20. [ Links ]
77. Kwak B, Mulhaupt F, Myit S, Mach F. Statins as a newly recognized type of immunomodulator. Nat Med 2000; 6: 1399-1402. [ Links ]
78. Kobashigawa JA, Katznelson S, Laks H, et al. Effect of pravastatin on outcomes after cardiac transplantation. N Engl J Med 1995; 333: 621-627. [ Links ]
79. Wenke K, Meiser B, Thiery J, et al. Simvastatin reduces graft vessel disease and mortality after heart transplantation: a four-year randomized trial. Circulation 1997; 96: 1398-1402. [ Links ]
80. Kwak B, Mulhaupt F, Myit S, et al. Statins as a newly recognized type of immunomodulator. Nature Med 2000; 6: 1399-1402. [ Links ]
81. Laurat E, Poirier B, Tupin E. In vivo down regulation of T helper cell 1 immune responses reduces atherogenesis in apolipoprotein E-knockout mice. Circulation 2001; 104: 197-202. [ Links ]
82. Takemoto M, Node K, Nakagami H, et al. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J Clin Invest 2001; 108: 1429-1437. [ Links ]
83. Buemi M, Senatore M, Corica F, et al. Statins and progressive renal disease. Med Res Rev 2002; 22: 76-83. [ Links ]
84. Arita S, Une S, Ohtsuka S, et al. Prevention of primary islet isograft nonfunction in mice with pravastatin. Transplantation 1998; 65: 1429-1433. [ Links ]
85. Freeman DJ, Norrie J, Sattar N, et al. Pravastatin and the development of diabetes mellitus: evidence for a protective treatment effect in the West of Scotland Coronary Prevention Study. Circulation 2001; 103:357-362. [ Links ]
86. Aronow WS, Ahn C, Kronzon I, Goldman ME. Association of coronary risk factors and use of statins with progression of mild valvular aortic stenosis in older persons. Am J Cardiol 2001; 88: 693- 695. [ Links ]
87. Olsson M, Thyberg J, Nilsson J. Presence of oxidized low density lipoprotein in non-rheumatic stenotic aortic valves. Arterioscler Thromb Vasc Biol 1999; 19: 1218-1222. [ Links ]
88. Mohler ER, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation 2001; 103: 1522-1528. [ Links ]
89. Bellamy MF, Pellikka PA, Klarich KW, Tajik AJ, Enriquez-Sarano M. Association of cholesterol levels, hydroxymethylglutaryl co-enzyme A reductase inhibitor treatment and progression of aortic stenosis. J Am Coll Cardiol 2002; 40: 1723-1730. [ Links ]
90. Rosenhek R, Rader F, Loho N, et al. Statins but not angiotensin converting enzyme inhibitors delay progression of aortic stenosis. Circulation 2004; 110: 1291-1295. [ Links ]
91. Wu B, Elmariah S, Kaplan FS, Cheng G, Mohler ER. Paradoxical effects of statins on aortic valve myofibroblasts and osteoblasts. Implications for end-stage valvular heart disease. Arterioscler Thromb Vasc Biol 2005; 25: 592-597. [ Links ]
92. Yamada M, Huang Z, Dalkara T et al. Endothelial nitric oxide synthase-dependent cerebral blood flow augmentation by l-arginine after chronic statin treatment. J Cereb Blood Flow Metab 2000; 20: 709-717. [ Links ]
93. Echeverri D, Montes F, Porto M, Moreno PR. Reducción del colesterol sérico y su repercusión en los efectos pleiotrópicos y morfológicos en placas ateroscleróticas tempranas en un modelo animal: implicaciones en el tratamiento y prevención de la aterosclerosis. Rev Col Cardiol 2004; 11 (1): 57-69. [ Links ]

