










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cardiología
Print version ISSN 0120-5633
Rev. Col. Cardiol. vol.15 no.1 Bogota Jan./Feb. 2008
(1) Universidad del Valle. Fundación Valle del Lili. Cali, Colombia.
(2) Clínica de Falla Cardiaca y Transplante. Fundación Valle del Lili. Cali, Colombia.
(3) Angiografía de Occidente SA. Cali, Colombia.
Correspondencia: Dr. Eduardo Contreras Zúñiga. Calle 4 No. 65 - 14. Celular: 317-5009197. Correo electrónico: edo11@hotmail.com. Fundación Valle del Lili, Cali, Colombia.
Recibido: 20/11/2007. Aceptado: 07/03/2008.
El síndrome de QT largo (SQTL) es una enfermedad que se caracteriza por la alteración electrocardiográfica en la repolarización ventricular que se manifiesta por prolongación del intervalo QT, secundaria a prolongación de la repolarización ventricular. Esto hace más vulnerables a dichos pacientes a arritmias ventriculares muy rápidas como «torsade des pointes» o fibrilación ventricular. El síndrome se observa generalmente en personas jóvenes y se asocia con riesgo de muerte súbita. Puede aparecer como parte del SQTL congénito (Jervell y Lange-Nielsen y Romano-Ward), o puede ser adquirido secundario a alteraciones metabólicas, tóxicas u otros factores fisiopatológicos.
Palabras clave: síndrome de QT largo, muerte súbita, trastornos de repolarización, arritmias.
Long QT syndrome is a disease characterized by the electrocardiographic alteration in ventricular repolarization manifested by prolonged QT interval, secondary to prolonged ventricular repolarization. This makes these patients more vulnerable to very fast ventricular arrhythmias such as torsade des pointes or ventricular fibrillation. This syndrome is generally observed in young people and is associated with sudden death. It may appear as part of congenital LQTS (Jervell and Lange-Nielsen and Romano-Ward), or may be secondarily acquired due to metabolic or toxic alterations or to other physiopathologic factors.
Key words: long QT syndrome, sudden death, repolarization disorders, arrhythmias.
El síndrome de QT largo es una alteración del sistema de conducción del corazón, que afecta un proceso denominado repolarización, que hace relación al restablecimiento de la carga eléctrica del corazón después de cada latido. El síndrome congénito de QT largo, es un trastorno poco común que generalmente se hereda. En otros casos, lo pueden ocasionar ciertos medicamentos o puede ser el resultado de un accidente cerebral vascular o de otro trastorno neurológico. El síndrome de QT largo puede producir arritmia, síncope e incluso muerte súbita (1, 2).
Etiología
Cuando el corazón se contrae, envía una señal eléctrica que se produce por el flujo de iones (moléculas de potasio, sodio y calcio) dentro de las células cardíacas. Los iones entran y salen de las células cardíacas a través de canales iónicos (2, 3).
Por medio de un electrocardiógrafo es posible registrar la señal eléctrica que emiten los iones. Esta máquina realiza un trazado de la señal, que se denomina «forma de onda». Las diferentes partes de la forma de onda se representan con las letras P, Q, R, S y T (3, 4).
Al observar la forma de onda, es posible determinar cuánto tiempo tarda la señal eléctrica en activar y desactivar las cavidades inferiores del corazón (ventrículos). Esto se denomina intervalo QT. Un problema en uno de los canales iónicos prolonga el intervalo QT, lo cual a su vez aumenta el riesgo de sufrir un tipo de arritmia denominado torsade de pointes (puntas torcidas). Cuando esto ocurre, el corazón no bombea suficiente sangre rica en oxígeno al resto del organismo, en especial al cerebro. La torsade de pointes también puede dar lugar a fibrilación ventricular, un tipo peligroso de arritmia que produce contracciones rápidas y no coordinadas de las fibras musculares de los ventrículos. La fibrilación ventricular impide que el corazón bombee sangre rica en oxígeno al resto del cuerpo, lo cual puede ocasionar la muerte (4-6).
Bases celulares
La dispersión del intervalo QT es un marcador de la heterogeneidad en la repolarización ventricular. Su aumento es un signo de inestabilidad eléctrica, que reduce el umbral para la fibrilación ventricular y facilita la aparición de arritmias ventriculares (4, 7).
Esta heterogeneidad secundaria a diferencias regionales del potencial de acción y los tiempos de activación que se demuestran en el eje apical basal y anterior posterior, entre el endocardio y el epicardio, también puede estar presente entre ambos ventrículos (interventricular), en un ventrículo (intraventricular), o en la pared ventricular (transmural). Algunos estudios indican gradientes significativos entre estas zonas, aunque el más importante es el transmural, en el cual existe heterogeneidad y, por tanto, mayor sustrato para el desarrollo de arritmias (2, 8, 9).
En condiciones normales, el potencial de acción más corto ocurre en el epicardio y el más largo en la región miocárdica (M). La duración del potencial de acción endocárdico es intermedia. En cuanto a la histología, las células M son similares a las epicárdicas y a las endocárdicas, pero sus características electrofisiológicas y farmacológicas se parecen a las de las células de Purkinje.
El potencial de acción de las células M se caracteriza por ser más prolongado cuando existen frecuencias cardiacas lentas o agentes que alarguen el potencial de acción. Las bases iónicas de este rasgo, están dadas por retraso o bloqueo de las corrientes de iones K+ y aumento de las corrientes de iones Na+ (3, 4, 8).
Cuando se bloquean los canales de K ocurre una prolongación preferencial del potencial de acción de las células M, que deriva en un marcado aumento de la dispersión transmural de la repolarización. Efectos similares ocurren con agentes que prolongan la duración del potencial de acción, como los bloqueadores de iones K+ (sotalol, eritromicina) y aquellos que aumentan el flujo de Na (9, 10).
Las diferencias regionales en la repolarización de las células M, son la razón fundamental de la dispersión del QT y esto se incrementa bajo condiciones de isquemia a causa de la amplificación de la heterogeneidad. La prolongación preferencial de la duración del potencial de acción de las células M que ocurre bajo estas condiciones, ocasiona un aumento paralelo de la dispersión transmural de la repolarización (10, 11).
Esta exagerada heterogeneidad transmural puede formar el sustrato para la reentrada y precipitar eventos que vencen la ventana vulnerable e inician la arritmia reentrante (8, 9).
Las diferencias regionales en la duración del potencial de acción de las células M, constituyen la base de la dispersión del QT medida en el electrocardiograma de superficie.
Después de la activación endocárdica, el epicardio es el último en despolarizarse, pero el primero en repolarizarse. La rápida repolarización del epicardio hace que la onda T desarrolle una polaridad similar a la del complejo QRS. El comienzo de la onda T guarda correspondencia con el declinar más rápido del plateau o fase 2 del potencial de acción epicárdico, y crea así un gradiente de voltaje entre el epicardio y la región M. El aumento de este gradiente es paralelo a medida que continúa la repolarización en el epicardio, hasta que se alcanza el máximo de amplitud, que marca el pico de la onda T. Entre el endocardio y la región M se desarrolla un gradiente similar pero opuesto, el cual limita su amplitud y constituye la rama descendente inicial de la onda T. La porción final de la rama descendente se debe a la disipación del gradiente por la repolarización de las células M. La total repolarización de esta región marca el final de la onda T. El intervalo entre el pico y el final de la T representa la dispersión transmural de la repolarización y puede ser un valioso índice electrocardiográfico (Figura 1) (11-13).
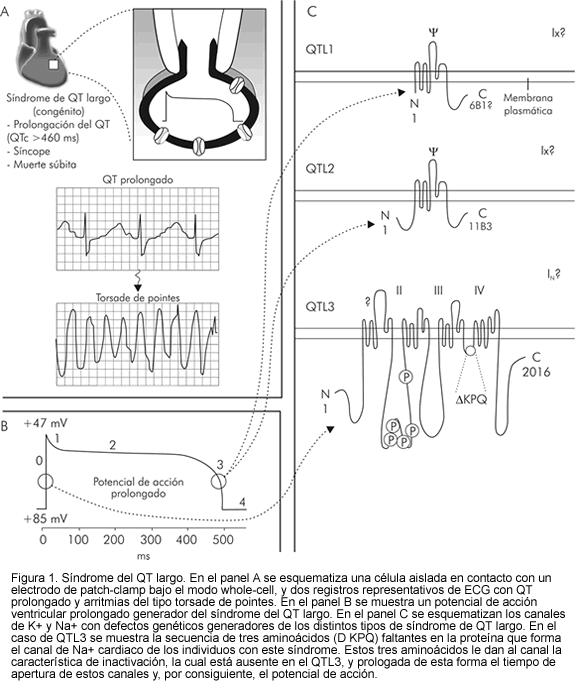
QT largo congénito
Los síndromes familiares de QT largo (QTL) son infrecuentes y se caracterizan por grupos familiares que tienen prolongación del intervalo QT en el electrocardiograma, mayor incidencia de síncope, taquicardia ventricular polimórfica del tipo torsade de pointes y muerte súbita. En el electrocardiograma se detecta repolarización prolongada y morfológicamente anormal, aunque no todos los pacientes afectados tienen tales alteraciones (13, 14).
Durante las últimas dos décadas se pensaba que ocurría por un disbalance fisio-patológico en la inervación autonómica del corazón. Estudios genéticos demostraron una alteración primaria en los canales que controlan las corrientes iónicas en la célula cardiaca como la responsable del cuadro. Al parecer, la activación del sistema nervioso simpático favorece la aparición de taquiarritmias ventriculares, sobre todo en pacientes portadores de anormalidades en el funcionamiento de los canales de potasio (15, 16).
En el síndrome de QT largo relacionado al cromosoma 3 (QTL 3), existe una alteración en el canal de sodio SCN5A que es responsable de la inactivación rápida de la corriente entrante de sodio. Esto motiva una corriente sódica hacia el interior celular durante el plateau del potencial de acción que puede inhibirse con lidocaína. Este síndrome es el más raro de los QTL. En estos pacientes, la activación adrenérgica no predispone al agravamiento del cuadro con la producción de arritmias ventriculares y torsade de pointes como ocurre en los pacientes con alteración en los canales de potasio. Se pueden beneficiar con marcapasos, ya que al evitar las bajas frecuencias se obvia la excesiva prolongación del QT (15, 17).
El síndrome de QT relacionado al cromosoma 7 (QTL 2) se caracteriza por una alteración en el gen HERG que codifica el canal de potasio para la Ikr (componente rápido de la delayed rectifier). En estos pacientes, el QT se prolonga ante la estimulación adrenérgica o ante el marcapasos, y se acorta con beta-bloqueadores y probablemente con el aumento de la concentración de potasio en suero (17).
De otra parte, el síndrome de QT relacionado al cromosoma 11 (QTL 1) fue el primero en descubrirse y es el más prevalente. Se trata de una alteración en las corrientes de potasio Iks.
QT Largo adquirido
En casos de isquemia miocárdica, hipertrofia, alteraciones hemodinámicas y electrolíticas se observan con frecuencia alteraciones del QT.
En pacientes con insufuciencia cardiaca ocurren ciertos cambios en corrientes iónicas en forma no homogénea. Esto crea heterogeneidad en la repolarización ventricular y las condiciones para la producción de arritmias cardiacas. En ratas portadoras de infarto crónico del miocardio, las células miocárdicas no infartadas tienen menor cantidad de ARNm que codifica la síntesis de proteínas para los canales de potasio. Los estados hemodinámicos alterados decodificarían genes que intervendrían en la repolarización ventricular y en la producción de arritmias cardiacas (16, 18, 19).
Muchos medicamentos antiarrítmicos prolongan el intervalo QT, lo cual lleva a la producción de proarritmia. Los niveles de potasio y los de calcio también alteran el QT.
Riesgo
El síndrome de QT largo puede afectar a quienes parecen encontrarse en muy buen estado de salud; en general, afecta a niños y adultos jóvenes. También existe mayor riesgo de padecer síndrome de QT largo si otros miembros de la familia sufren este trastorno.
En algunos casos, los medicamentos antiarrítmicos y los antidepresivos también aumentan el riesgo de padecer este síndrome (19, 20).
Síntomas
En general, los pacientes con síndrome de QT largo son asintomáticos; cuando se dan, los más comunes son el desmayo y la arritmia. Así mismo, a menudo presentan un intervalo QT prolongado durante el ejercicio físico, en momentos de emoción intensa (por ejemplo: temor, ira o dolor) o en reacción a un sonido fuerte o alarmante (17, 21).
Estos pacientes han tenido por lo menos un episodio de desmayo antes de cumplir los diez años de edad. Otros pueden tener sólo uno o dos episodios de desmayo en la niñez y de ahí en adelante no tener episodios adicionales (22, 23).
La sordera es un síntoma que corresponde a un tipo de síndrome de QT heredado.
Diagnóstico
El síndrome de QT largo se diagnostica mediante las siguientes técnicas:
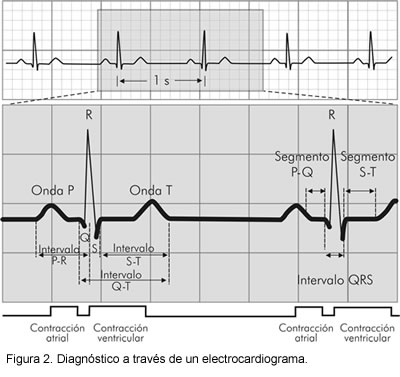
- Electrocardiograma convencional: es el mejor estudio para diagnosticar el síndrome de QT largo. El electrocardiógrafo registra la actividad eléctrica del corazón en formas de onda que pueden indicar un intervalo QT prolongado (Figura 2).
- Electrocardiograma de esfuerzo: también se denomina prueba de esfuerzo y puede mostrar un intervalo QTc anormal que un electrocardiograma en reposo posiblemente no detecte (Figura 3).

- Estudio Holter: ofrece una lectura continua de la frecuencia y el ritmo cardiaco durante un período de 24 horas (o más). El paciente lleva puesto un dispositivo de grabación (el monitor Holter) que se conecta a pequeños electrodos que se ubican sobre el tórax. Luego, los médicos pueden estudiar el registro impreso de la grabación para determinar si se produjo un intervalo QT prolongado (23, 24).
Algunas personas con síndrome de QT largo pueden no tener un intervalo QT prolongado todo el tiempo y, por consiguiente, no siempre se descubre el trastorno durante un chequeo de rutina; de ahí la importancia de conocer los antecedentes médicos familiares. En toda familia en que se produzcan varios episodios de desmayo o que tenga antecedentes de muerte súbita, el causante podría ser el síndrome de QT largo (25).
Tratamiento
A futuro será posible realizar análisis genéticos a los pacientes portadores de QT largo y a sus familiares, lo que permitirá estudiar el tipo de mutación genética y la existencia de portadores sanos en la familia, y probablemente se podrá adecuar la terapéutica según la anormalidad hallada. Hoy en día estos análisis son dispendiosos, largos y costosos, y por otra parte, no se dispone de alternativas terapéuticas superiores a los beta-bloqueadores. El tratamiento del síndrome de QT largo puede incluir cambios en el estilo de vida, medicamentos o cirugía (25, 26).
Cambios en el estilo de vida
A menudo, tras comenzar el tratamiento, los pacientes con síndrome de QT largo pueden participar en deportes recreativos y otras actividades, siempre y cuando lo hagan con moderación. Si tienen episodios de desmayo al hacer ejercicio, es aconsejable la compañía de un amigo o familiar que esté alerta en conseguir asistencia si se requiere.
Al día de hoy se dispone de cinco modalidades terapéuticas:
1. Beta-bloqueadores.
2. Terapéutica farmacológica específica según la alteración genética.
3. Marcapasos.
4. Simpaticectomía simpática cervicotorácica izquierda.
5. Cardiodesfibrilador automático implantable.
El objetivo del tratamiento es la prevención de las arritmias ventriculares malignas y la muerte súbita (26, 27).
Medicamentos beta-adrenérgicos
Se utilizan en quienes ya presentaron un episodio de síncope o muerte súbita abortada o en asintomáticos con historia familiar de muerte súbita. Son muy útiles para los trastornos con alteración de la corriente saliente de potasio «delayer rectifier» que es muy dependiente de la actividad adrenérgica. La efectividad en el tratamiento es menos clara en los pacientes que tienen alteración del canal de sodio SCN5A (LQT3) (28, 29).
Terapéutica farmacológica específica según la alteración genética
El tratamiento con mexiletina por vía oral, acorta el QT y normaliza la morfología de la onda T en los pacientes con LQT3. La infusión de potasio con espironolactona por vía oral, corrige las anormalidades de la repolarización en los LQT2. Hasta el momento no existen seguimientos a largo plazo con estas terapias (29, 30).
Marcapasos
La terapéutica combinada de marcapasos y beta-bloqueadores ha demostrado gran efectividad en el tratamiento de pacientes con arritmias malignas recurrentes. Probablemente, al prevenir las pausas el marcapasos evita el desencadenamiento de arritmias pauso-dependientes (27, 31).
Gangliectomía cervicotorácica simpática izquierda
La eficacia de este tratamiento es controvertida y se reserva para pacientes refractarios a los beta-bloqueadores y al marcapasos.
Cardiodesfibrilador automático implantable
Se recomienda en sobrevivientes de muerte súbita o en los casos refractarios al tratamiento farmacológico. Con el avance tecnológico los dispositivos son cada vez más pequeños, lo que los hace sencillos de implantar en niños. De igual forma, la capacidad de programación hace que puedan emitir descargas solamente cuando exista una arritmia letal y no tratar episodios cortos de torsade de pointes que llevarían a un agotamiento precoz del aparato (18, 30, 31).
Bibliografía
1. Madeiros A. Clinical and genetic characteristics of long QT syndrome. Rev Esp Cardiol 2007; 60 (7): 739-52. [ Links ]
2. Goldenberg I. Sudden cardiac death without structural heart disease: update on the long QT and Brugada syndromes. Curr Cardiol Rep 2005; 7 (5): 349-56. [ Links ]
3. Priori S. New insights into the long-QT syndrome. Rev Esp Cardiol 2007; 60 (7): 675-82. [ Links ]
4. Leroy S. Long QT syndrome and other repolarization-related dysrhythmias. AACN Clin Issues 2004; 15 (3): 419-31. [ Links ]
5. Madeiros A. New perspectives in long QT syndrome. Rev Invest Clin 2007; 59 (1): 57-72. [ Links ]
6. Brugada J. Channelopathies: a new category of diseases causing sudden death. Herz 2007; 32 (3): 185-91. [ Links ]
7. Chiang C. Congenital and acquired long QT syndrome. Current concepts and management. Cardiol Rev 2004; 12 (4): 222-34. [ Links ]
8. Bar E. Guidelines for treating cardiac manifestations of organophosphates poisoning with special emphasis on long QT and torsades de pointes. Crit Rev Toxicol 2007; 37 (3): 279-85. [ Links ]
9. Darbar B. Pharmacogenetics of antiarrhythmic therapy. Expert Opin Pharmacother 2006; 7 (12): 1583-90. [ Links ]
10. Nader A. Inherited arrhythmic disorders: long QT and Brugada syndromes. Tex Heart Inst J 2007; 34 (1): 67-75. [ Links ]
11. Santana LF. Sodium current and arrhythmogenesis in heart failure. Heart Fail Clin 2005; 1 (2): 193-205. [ Links ]
12. Ching C. Congenital long QT syndromes: clinical features, molecular genetics and genetic testing. Expert Rev Mol Diagn 2006; 6 (3): 365-74. [ Links ]
13. Celi A. QT dispersion: time for a revival? Intern Emerg Med 2006; 1 (4): 262-3. [ Links ]
14. Borchert D. Long and short QT syndrome. Herzschrittmacherther Elektrophysiol 2006; 17 (4): 205-10. [ Links ]
15. Ingles J. Sudden cardiac death in the young: a clinical genetic approach. Intern Med J 2007; 37 (1): 32-7. [ Links ]
16. Napolitano C. Long QT syndrome and short QT syndrome: how to make correct diagnosis and what about eligibility for sports activity. J Cardiovasc Med (Hagerstown) 2006; 7 (4): 250-6. [ Links ]
17. Skinner JR. Guidelines for the diagnosis and management of familial long QT syndrome. Heart Lung Circ 2007; 16 (1): 22-4. [ Links ]
18. Roden DM. Drug-induced long QT and torsade de pointes: recent advances. Curr Opin Cardiol 2007; 22 (1): 39-43. [ Links ]
19. Collins K. Advances in congenital long QT syndrome. Curr Opin Pediatr 2006; 18 (5): 497-502. [ Links ]
20. Bernal O. Cardiac arrhythmias in women. Rev Esp Cardiol 2006; 59 (6): 609-18. [ Links ]
21. Tan HL. Sodium channel variants in heart disease: expanding horizons. J Cardiovasc Electrophysiol 2006; 17 (suppl 1): S151-S157. [ Links ]
22. Hondeghem D. Thorough QT/QTc not so thorough: removes torsadogenic predictors from the T-wave, incriminates safe drugs, and misses profibrillatory drugs. J Cardiovasc Electrophysiol 2006; 17 (3): 337-40. [ Links ]
23. Jalaie M. QT interval prolongation: and the beat goes on. Mini Rev Med Chem 2005; 5 (12): 1083-91. [ Links ]
24. Anderson ME. QT interval prolongation and arrhythmia: an unbreakable connection? J Intern Med 2006; 259 (1): 81-90. [ Links ]
25. Schwartz P. The congenital long QT syndromes from genotype to phenotype: clinical implications. J Intern Med 2006; 259 (1): 39-47. [ Links ]
26. Schwartz P. Management of long QT syndrome. Nat Clin Pract Cardiovasc Med 2005; 2 (7): 346-51. [ Links ]
27. Shimizu W. The long QT syndrome: therapeutic implications of a genetic diagnosis. Cardiovasc Res 2005; 67 (3): 347-56. [ Links ]
28. Gupta A. Current concepts in the mechanisms and management of drug-induced QT prolongation and torsade de pointes. Am Heart J 2007; 153 (6): 891-9. [ Links ]
29. Khan I. Novel therapeutics for treatment of long-QT syndrome and torsade de pointes. Int J Cardiol 2004; 95 (1): 1-6. [ Links ]
30. Zabera W. Role of implantable cardioverter defibrillator therapy in patients with long QT syndrome. Am Heart J 2007; 153 (4 Suppl): 53-8. [ Links ]
31. Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med 2004; 350 (10): 1013-22. [ Links ]

