











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
El síndrome de microdeleción 22q11 (SD22q11), es causado por una deleción hemicigótica del brazo largo del cromosoma 22, la cual fue descrita por primera vez en 1965 por Angelo DiGeorge. Es el síndrome de microdeleción más común en el mundo, con una prevalencia de 1 en 4000-6000 nacidos vivos, y aproximadamente en 1 de cada 1000 fetos; sin embargo, puede haber mayor prevalencia de casos que son subdiagnosticados por la amplia variabilidad en la expresión clínica. El diagnóstico en el SD22q11 puede darse al nacimiento o puede ser más tardío, durante la infancia o incluso en la adolescencia1. Alrededor del 70 al 80% de los casos se presentan con alguna cardiopatía congénita; la más común de ellas, en un 20%, es la tetralogía de Fallot. El 3 al 6% de estos pacientes presentan síndrome de agenesia de la válvula pulmonar concomitante; otras cardiopatías congénitas que se observan son la comunicación interventricular (CIV), el tronco arterioso, el arco aórtico derecho y otras anomalías del arco aórtico2,3. En el 90% de los casos, la alteración cromosómica es ocasionada por una deleción de novo y en el otro 10% se transmite como un rasgo autosómico dominante2,4, lo cual significa que el 50% de los hijos de las personas con la alteración cromosómica tienen la probabilidad de tener un hijo afectado durante cada embarazo5. Históricamente, diversos síndromes, como el de DiGeorge, el velocardiofacial o de Shprintzen; el de anomalía conotruncal de la cara y el de Opitz G/BBB autosómico dominante, han sido descritos por separado6. Sin embargo, a través del análisis citogenético y molecular mediante la técnica de hibridación fluorescente in situ (FISH), se descubrió que la causa genética común en el 90% de estos pacientes fue la microdeleción del cromosoma 22q111.
De acuerdo con el grupo de edad hay características clínicas, como cardiopatías congénitas, anomalías del paladar, rasgos faciales típicos e inmunodeficiencia como consecuencia de aplasia e hipoplasia tímica1, que hacen sospechar en una deleción 22q11.
El tratamiento del SD22q11 va dirigido, en primer lugar, a salvar la vida del paciente, solventando la urgencia en la forma de presentación y, posteriormente, tratando de mejorar la calidad de vida.
El objetivo de este manuscrito es reportar un caso clínico de SD22q11 asociado a tetralogía de Fallot con síndrome de agenesia valvular pulmonar en una recién nacida de quince días de vida, atendida en el Hospital Vicente Corral Moscoso, Cuenca, Ecuador. Para ello se siguieron los lineamientos CARE para el reporte de casos clínicos.
Caso clínico
Se trata de una recién nacida de sexo femenino, de quince días de vida, procedente de Chaucha, provincia de Azuay, Ecuador. Como antecedentes de importancia destacan madre de 29 años de edad, G5P5HV5, con cinco controles prenatales y consumo de hierro y ácido fólico a partir del quinto mes del embarazo; como antecedente de riesgo refiere vaginosis al sexto mes de gestación, tratada con óvulos vaginales.
Recién nacida viva única que nace por parto vaginal espontáneo, con un peso de 2710 gramos, talla de 47 cm, perímetro cefálico de 33 cm (desviación estándar: -1, -2) y Ballard de 38 semanas de gestación. Las puntuaciones del APGAR fueron de 5 y de 7 al primer y décimo minuto de vida, respectivamente. Durante el primer minuto presentó flacidez, hipotonía con pobre esfuerzo respiratorio, palidez generalizada y frecuencia cardíaca menor de 100 lpm, por lo que se inició ventilación con presión positiva durante dos ciclos. Al permanecer con una frecuencia cardíaca menor de 100 lpm, palidez y pobre esfuerzo respiratorio, se decidió intubación endotraqueal con lo cual mejoró la frecuencia cardíaca, pero persistió el pobre esfuerzo respiratorio con una mala mecánica ventilatoria, retracciones intercostales y una saturación de oxígeno > 90%. Durante tres episodios presentó desviación de la mirada y movimientos tónicos, por lo que se decidió su ingreso a la unidad de neonatología.
Al examen físico, se pudo evidenciar, en cabeza, hipoplasia malar, nariz bulbosa, puente nasal ancho, paladar íntegro, llanto débil, orejas de implantación baja y micrognatia. Campos pulmonares: ventilados con murmullo vesicular conservado; en corazón: soplo sistodiastólico de intensidad II/IV en focos de base y tricúspide; en columna: mechón de pelo en la región sacra, sugerente de espina bífida oculta. Al examen neurológico, se observó alerta, con reflejos de succión y plantar presentes. En las extremidades se evidenció polidactilia axial en el pie izquierdo (Fig. 1).
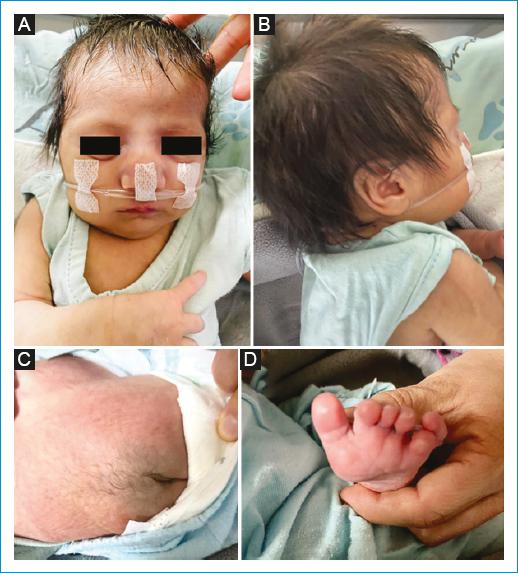
Figura 1 Manifestaciones clínicas. A: base nasal ancha, hipoplasia malar, micrognatia. B: orejas de implantación baja. C: mechón de pelo en la región sacra de la columna vertebral, sugerente de espina bífida oculta. D: polidactilia axial en el pie izquierdo. [Cuenca, Azuay, Ecuador; 17/01/2023].
En la analítica que se realizó durante el ingreso, se halló hipoproteinemia (5.9 g/dl), elevación de LDH (704 U/L), hipercalcemia (12 mg/dl), elevación de CK-MB (142.1 U/l), CPK creatina-fosfocinasa (1809 U/L) y troponina T (51.4 pg/ml).
La ecografía transfontanelar, renal y abdominal fue normal. En la ecografía del canal medular se evidenciaron hallazgos sugestivos de lesión de posible origen quístico a nivel del canal medular distal.
Se le realizó un ecocardiograma transtorácico que evidenció función biventricular normal (FEVI del 87%); comunicación interauricular (CIA) tipo foramen oval de 3 mm, tetralogía de Fallot con defecto interventricular subaórtico de 5 mm, a nivel del anillo pulmonar (z score: 0), anillo estenótico con ausencia de valvas sigmoideas e insuficiencia pulmonar grave, y dilatación de la rama derecha (z score: +2.7) e izquierda (z score: +3.9) de la arteria pulmonar. Se observó, además, presencia de arco aórtico derecho. Se solicitó una angiotomografía cardíaca en la que se confirmaron los hallazgos ecocardiográficos, dilatación de las ramas de la arteria pulmonar, tetralogía de Fallot más arco aórtico derecho y broncomalacia (Fig. 2).
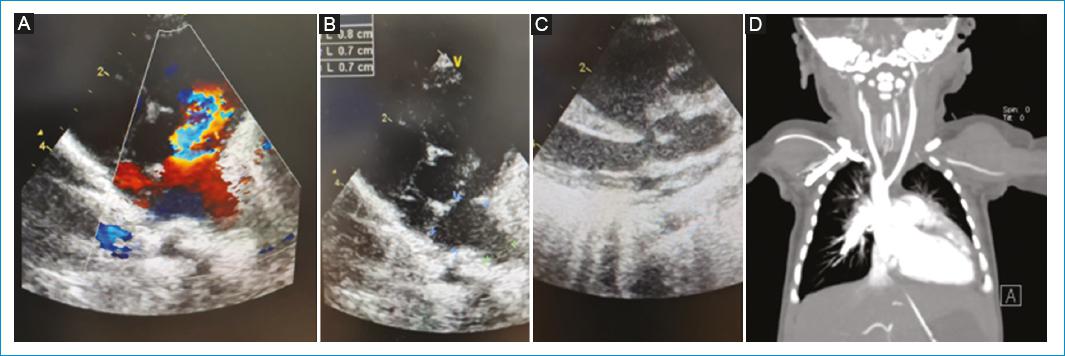
Figura 2 Hallazgos ecocardiográficos y angiotomográficos. A: se observa el anillo de la válvula pulmonar con dilatación aneurismática de las ramas de la arteria pulmonar y aliasing doppler a nivel anular. B: dilatación de las ramas de la arteria pulmonar, rama derecha de 7 mm y rama izquierda de 6 mm. C: anillo aórtico cabalgando el septo interventricular. D: incremento del tamaño de las cavidades cardíacas derechas con estenosis pulmonar; el tronco principal de la arteria pulmonar de 10.5 mm de tamaño, su rama derecha de 11.4 mm y su rama izquierda de 9.6 mm. Comunicación interauricular (CIA) de 4 mm de diámetro. Cabalgamiento de la aorta sobre el septum interventricular. Arco aórtico derecho con arteria subclavia aberrante del lado izquierdo [Servicio de Imagenología y Radiología del Hospital «Vicente Corral Moscoso». Cuenca, Azuay, Ecuador; 04/01/2023].
Con los datos clínicos y las características imagenológicas se llegó a la sospecha de SD22q11, por lo que se efectuó una prueba de hibridación fluorescente in situ (FISH) que confirmó la microdeleción del cromosoma 22q11 y, por tanto, la sospecha clínica (Fig. 3).
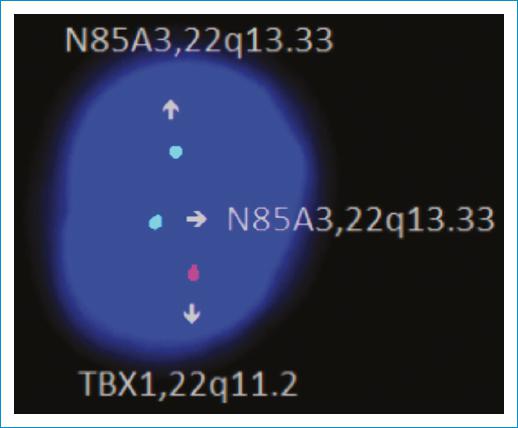
Figura 3 Prueba de hibridación fluorescente in situ (FISH): se observa una señal para el gen TBX1 y dos señales para el gen N85A3, compatible con una deleción de la región 22q11.2 en 25 células analizadas. Este resultado es consistente con el diagnóstico de síndrome de DiGeorge [Cuenca, Azuay, Ecuador; 11/01/2023].
Discusión
Las cardiopatías congénitas tienen una frecuencia de 4 a 50 por cada 1000 nacidos vivos en el mundo, las cuales pueden tener asociación con alteraciones genéticas, y de estas el 5 al 10% estarán causadas por anomalías cromosómicas, en especial la trisomía de los cromosomas 13, 18 y 21. En un 25% de los casos, la intervención inmediata deberá ser antes del primer año de vida ya que puede progresar a cuadros de shock cardiogénico, cianosis y edema pulmonar7. En Ecuador, se ha determinado que la prevalencia de cardiopatías congénitas en neonatos es de 2.7 por cada 1000 nacidos vivos; en la provincia del Azuay y en la ciudad de Cuenca, se determinaron tasas de incidencia de 2.35 y 2.74 por cada 1000 nacidos vivos, respectivamente8.
La tetralogía de Fallot y su asociación con el síndrome de agenesia valvular pulmonar es una rara variante que se presenta en el 3 al 6% de estos pacientes; en esta, a nivel del anillo valvular, existen valvas rudimentarias y un anillo estenosado, lo que conlleva dilatación aneurismática de la arteria pulmonar y sus ramas, que comprime la porción anterior del extremo inferior de la tráquea y los bronquios durante el desarrollo de la vida fetal, produce hipoplasia de las vías respiratorias comprimidas y, en consecuencia, insuficiencia respiratoria, la cual es la principal causa de mortalidad de estos pacientes4,9.
En este reporte se presenta el caso clínico de una paciente recién nacida con características propias del SD22q11, tanto clínicas como imagenológicas. Las manifestaciones clínicas más tempranas fueron insuficiencia respiratoria aguda, palidez generalizada, hipotonía y bradicardia; adicionalmente, durante el examen físico, se evidenciaron orejas de implantación baja, hipoplasia malar, micrognatia, puente nasal ancho, llanto débil, soplo cardíaco y polidactilia axial en el pie izquierdo. Desde el punto de vista radiológico, los hallazgos ecocardiográficos y de la angiotomografía cardíaca, se asociaron con los de una cardiopatía congénita compleja, por lo que se realizó el cribado de SD22q11. Dentro del espectro de hallazgos clínicos, una posible espina bífida oculta por la presencia de pelo en la región sacra y el hallazgo ecográfico de lesión quística en el canal medular, probablemente fueron consecuencia de patologías concomitantes. Como se menciona en la guía de la Asociación Americana del Corazón (AHA) de cardiopatías congénitas, fue necesario realizar el cribado de SD22q11 ante el hallazgo de cardiopatía congénita compleja; en este caso se realizó mediante técnica FISH y fue diagnosticada una microdeleción 22q1110.
En la deleción 22q11 existe una alteración en la región que contiene al gen TBX1, el cual es uno de los factores de transcripción implicado en el desarrollo del tercer y cuarto arcos faríngeos, que causan el desarrollo anormal del timo y de la paratiroides, la ausencia de características como la aplasia/hipoplasia del timo, hipoparatiroidismo con hipocalcemia y reflujo nasofaríngeo en un recién nacido es rara ya que por lo general son las principales características clínicas que hacen sospechar en el diagnóstico y esto tiene que ver con la amplia variabilidad en la expresión de los genes que forman parte de la región 22q11, por ello en este caso la sospecha clínica inicia a partir de las expresiones fenotípicas y de la cardiopatía congénita compleja10.
El tratamiento de la cardiopatía en los pacientes con SD22q11 ha generado un gran debate, pero se concuerda en que se debe realizar un diagnóstico rápido y oportuno debido a que el tratamiento definitivo de la cardiopatía congénita es quirúrgico. La reparación de la tetralogía de Fallot debe realizarse de acuerdo con la condición clínica y anatómica de cada paciente, por ello se sugiere que se realice antes del primer año, entre los tres a seis meses de vida11,12.
De acuerdo con la literatura, el tratamiento de pacientes con tetralogía de Fallot y agenesia de la válvula pulmonar consiste en la reparación primaria completa de la CIV mediante ventriculotomía derecha (a través del anillo pulmonar) y reemplazo de la válvula pulmonar displásica con un homoinjerto o por medio de un conducto con una válvula, y, así mismo, a nivel de la arteria pulmonar y sus ramas, pues, de esta forma, se reducen los efectos adversos de la hipoxia tisular y disminuye la evolución de la fibrosis y la hipertrofia del ventrículo derecho, optimizando la función ventricular a largo plazo. Existen cirujanos que plantean la sección aórtica para lograr una correcta exposición de la arteria pulmonar y así realizar una amplia arterioplastia pulmonar en ambos hilios pulmonares. La mortalidad quirúrgica es precoz y es superior al 20%; la tasa de supervivencia al año es del 75%12,13.
Conclusión
La principal alteración cardíaca en el SD22q11 es la tetralogía de Fallot, y su asociación con el síndrome de agenesia valvular pulmonar es rara, pero altamente sugestiva de SD22q11, por lo que ante estos hallazgos se debe realizar un cribado completo de alteraciones genéticas en un recién nacido.
Es posible diagnosticar el SD22q11 a partir de la observación fenotípica de elementos característicos en los recién nacidos; en este sentido, las anomalías faciales y las alteraciones cardíacas fueron elementos clave, que guiaron al equipo médico en la sospecha diagnóstica de este síndrome. Finalmente, las técnicas citogenéticas y moleculares, así como los hallazgos imagenológicos, fueron fundamentales en el diagnóstico definitivo del SD22q11.

