











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
Los glioblastomas son el tumor más común y maligno entre las neoplasias de la glía 1. Según el Registro Central de Tumores Cerebrales de los Estados Unidos (CBTRUS), la tasa de incidencia anual ajustada por edad de los tumores cerebrales primarios malignos es 7,06/100.000 habitantes y el glioblastoma fue el 49,1% de ellos 2. Sé caracteriza por ser un tumor de crecimiento rápido, compuesto por una mezcla heterogénea de células tumorales astrocitarias pobremente diferenciadas, con pleomorfismo, necrosis y proliferación vascular. Según la quinta clasificación de la Organización Mundial de la Salud este tipo de tumores no presenta mutación en el gen IDH y usualmente presenta mutación en el gen EGFR, mutación del promotor de TERT, trisomía del cromosoma 7 y monosomía del cromosoma 10 3. Puede manifestarse a cualquier edad, pero afecta principalmente a adultos, con un pico de incidencia entre los 45 y los 70 años 3.
En este tipo de neoplasias se han identificado diferentes genes que están implicados en el diagnóstico, algunos de ellos en el pronóstico y otros pocos con poder predictivo en la respuesta. Dentro de ellos juegan un papel muy importante, los receptores Tirosina Quinasa (RTQ), los cuales se caracterizan por ser reguladores centrales de vías de proliferación, diferenciación y apoptosis celular. La no regulación de la señalización mediada por estas proteínas se encuentra involucrada en la patogénesis de múltiples neoplasias como los son los glioblastomas 4.
Los RTQ son proteínas transmembrana que catalizan la fosforilación de los residuos de tirosina en las proteínas; la fosforilación de estos residuos influye en tres aspectos fundamentales: su actividad, localización y sus interacciones 5. Estas enzimas se encuentran en diferentes cascadas de señalización que van desde la membrana celular hasta el ADN del núcleo por lo que la alteración de las RTQ puede contribuir en la iniciación o en la progresión del cáncer 6. De la estructura de estos receptores se puede decir que están compuestos de 3 partes: 1) una porción extracelular que se une al ligando, 2) una porción transmembrana y 3) una porción intracelular con su dominio catalítico que es capaz de unirse y fosforilar un sustrato determinado. Cuando el ligando se une al receptor se producen cambios estructurales que permiten el contacto del adenosín trifosfato (ATP) con el sustrato. Luego se genera una cascada de señales a través del citoplasma que causa una respuesta en la expresión génica a nivel citoplasmático y nuclear. Muchos RTQ están involucrados en la oncogénesis ya sea por mutación del ADN, translocación cromosómica o una sobreexpresión génica 6.
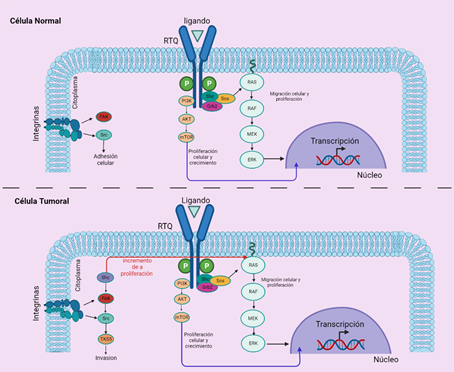
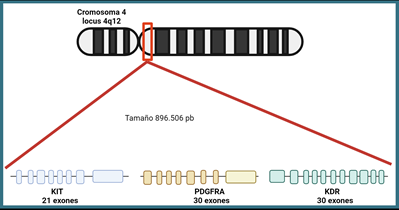
Estas moléculas actúan como receptores de factores de crecimiento, hormonas, citoquinas, factores neurotróficos y otras moléculas de señalización extracelular. Tras la activación por ligandos, los RTQ envían señales a través de dos vías principales las cuales se encuentran localizadas en la región UTR 3´; estas vías son Ras/MAPK/ERK y Ras/PI3K/AKT, las cuales se pueden ver alteradas en las células con cáncer (Figura 1) 7. Estas cascadas de señalización están implicadas en la regulación de la proliferación, supervivencia, diferenciación y angiogénesis celular. Este reporte de caso se enfoca en tres RTQ: el receptor A del factor de crecimiento derivado de las plaquetas (PDGFRA) codificado por el gen PDGFRA para su ligando factor de crecimiento derivado de las plaquetas (PDGF); el receptor del factor de células madre (c-Kit o CD117) codificado por el gen KIT para su ligando factor de crecimiento de células madre (SCF) y el receptor 2 del factor de crecimiento endotelial vascular (VEGFR-2), codificado por el gen KDR para su ligando, el factor de crecimiento del endotelio vascular (VEGF), estando los tres genes ubicados en el brazo largo del cromosoma 4 (4q11-q12) (Figura 2) 8. En este estudio se investigó la relación entre estos oncogenes evaluados de forma rutinaria en un panel de 324 genes 9 junto con las características clínicas, morfológicas y moleculares de los pacientes con gliomas de alto grado; además, se describe como estas variantes genéticas podrían tener un significado pronóstico, así como potencial relevancia para el tratamiento.
En las células normales, la unión de ligandos (o señales de adhesión mediadas por integrinas) induce la dimerización del receptor y la fosforilación de la tirosina. Los RTQ activados, a su vez, inician las cascadas de señalización de las vías RAS/MAPK, PI3K/Akt/mTOR o FAK/Src mediante el reclutamiento de proteínas adaptadoras Grb2 y Shc. Esto da como resultado respuestas celulares como la supervivencia, el crecimiento, la proliferación, la diferenciación y la migración. En las células cancerosas, la amplificación de RTK pueden dar lugar a un fenotipo oncogénico mediante la activación de vías de invasión, así como la proliferación descontrolada. Imagen tomada y modificada de Tilak et. al 2021 14.
Presentación de los casos
Este estudio fue de tipo observacional y de cohorte ambispectivo, debido a que se tomaron retrospectivamente los datos sociodemográficos y clínicos, mientras que los datos genómicos se obtuvieron prospectivamente. La población de estudio fueron pacientes que acudieron al Instituto de Cancerología de la Clínica Las Américas-AUNA entre los años 2019 y 2020, para que se les brindara tratamiento para gliomas de alto grado. De la población total se tomó una muestra de 31 pacientes por invitación abierta con diagnóstico de glioblastoma, oligodendroglioma y astrocitoma. La secuenciación genómica se realizó a través del panel FoundationOne®CDx (F1CDx) 9 que es una metodología de diagnóstico in vitro basado en secuenciación de próxima generación para la detección de mutaciones como: sustituciones, inserciones, alteraciones en el número de copias (CNA) en 324 genes y reordenamientos de genes seleccionados, así como huellas genómicas que incluyen inestabilidad de microsatélites (MSI), carga mutacional del tumor (TMB) y, puntuación de pérdida de heterocigosis (LOH). Los criterios de inclusión fueron: pacientes mayores de 18 años, diagnosticados con tumor cerebral maligno primario a los que se realizó cirugía de resección tumoral o biopsia, ellos deberían contar con el diagnóstico histológico de glioblastoma. Adicionalmente los pacientes debían tener mutaciones en los siguientes tres genes PDGFRA, KIT y KDR o la combinación de estos; es decir se podría encontrar al menos dos de estos tres genes mutados.
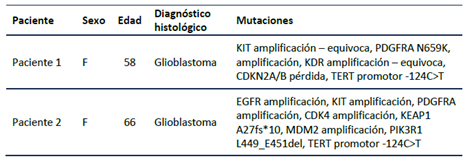
De la cohorte de 31 pacientes con glioma de alto grado, a quienes se les realizó F1CDx, se seleccionaron dos pacientes con diagnóstico de glioblastoma según la quinta edición de la clasificación de la Organización Mundial de la Salud 2021 con sobrevida menor de los 14,6 meses 10, cuyas características epidemiológicas y genómicas se muestran en la Tabla 1.
La primera es una mujer de 58 años que consulta por cefalea que aumenta con valsalva, la despierta en horas de la madrugada y aumenta con los movimientos de la cabeza. Narra además vomito no precedido de náuseas y oscuraciones. Al Examen físico se encuentra papiledema y déficit motor de hemicuerpo izquierdo. En la RM se documenta lesión temporal derecha. Se lleva a resección quirúrgica de un 60% (para fines prácticos se toma como equivalente a biopsia) y a las cuatro semanas se inicia protocolo Stupp con radioterapia y quimioterapia con temozolomida concomitante adyuvante de la cual solo recibe cuatro ciclos ya que presenta nuevamente cefalea con características de hipertensión endocraneana y hemiparesia izquierda densa. Se presenta en Junta de neuro-oncología en que se descarta nueva cirugía o re-irradiación y se inicia bevacizumab con poca respuesta.
La segunda es una mujer de 66 años que consulta por cefalea y crisis parciales simples motoras derechas que generalizan. Al examen físico se documenta perdida del pulso venoso y déficit sutil motor de hemicuerpo derecho. En la resonancia magnética (RM) se encuentra lesión parietal izquierda. Se hace una biopsia estereotáxica ya que la lesión estaba en un área elocuente y no permitía una buena citoreducción quirúrgica y a las 3 semanas se inicia protocolo Stupp, recibiendo solo cinco de los seis ciclos de adyuvancia programados ya que presenta un brusco deterioro clínico, ante lo cual se decide ofrecer solo tratamiento de soporte con esteroides.
Las dos pacientes presentaron un curso más tórpido que el resto del grupo, con síntomas de progresión a los 4 y 6 ciclos de adyuvancia respectivamente, lo cual se corroboro imagenlógicamente. La mediana de sobre vida libre de progresión (SLP) de la primera fue de 6,5 meses y de la segunda de 6,6 meses (siendo la mediana de SLP de 12 meses de los 31 pacientes incluidos con glioma de alto grado) y la sobrevida global (OS) de la primera fue de 12 meses y de la segunda de 14 meses (siendo la OS del grupo de los 31 pacientes con HGG de 19 meses), un poco menores que la cohorte y lo reportado por la literatura 10.
Como se observa en la Tabla 1, para el paciente 1 se reportaron las mutaciones en los genes de interés, esto es, PDGFRA, KID y KDR, presentando además mutación de CDKN2A/B y TERT. Si bien el paciente 2 no cumplió con la regla de tener mutaciones en los tres genes ya que este presentó solo mutaciones en los genes KIT y PDGFRA, se tenía como hallazgo interesante una variación en otro RTQ como lo es EGFR, adicionalmente se evidencio otras mutaciones en los genes CDK4, KEAP1, MDM2, PIK3R1 y TERT.
Consideraciones éticas
Como requisito del proceso de evaluación y uso de datos se obtuvo el consentimiento informado por parte de los participantes, además este estudio cuenta como aval del comité de ética de la Universidad CES aprobado en sesión 122 del 08 de mayo 2018.
Discusión
La amplificación en el locus 4q12 que alberga los genes KIT, KDR y PDGFRA, ocurre con una frecuencia del 15% en los glioblastoma 11. Es de anotar que nuestra cohorte de pacientes con glioma de alto grado se dividía en: 21 (68%) tenían glioblastoma, 5 (16%) tenían astrocitoma anaplásico, 4 (13 %) tenían oligodendroglioma anaplásico y 1 paciente tenía oligoastrocitoma, reportes que se reevaluaron al confrontarlos con el F1CDx, dos pacientes con glioblastoma fueron reclasificados a astrocitoma anaplásico y un oligodendroglioma anaplásico a astrocitoma anaplásico, en este caso las dos pacientes con diagnóstico de glioblastoma que poseían la mutación en el locus 4q12 representan un 6,5% del total de los pacientes, a pesar de que es un valor menor al reportado por la literatura este puede evidenciar la importancia de realizar el perfilamiento genético para identificar posibles dianas tanto pronósticas como terapéuticas, como es el caso de la amplificación del gen EGFR, la cual está relacionada con un peor pronóstico para pacientes con glioblastomas 12,31, es de resaltar que este porcentaje puede variar debido a las técnicas moleculares implementadas para la detección de las mutaciones 13.
El cambio producido por la mutación en estos tres genes aún no se ha descrito a nivel biológico y se desconoce su capacidad oncogénica 12. Esta variable generalmente se correlaciona con altos niveles de transcripción, que puede dar como resultado un aumento de los niveles de proteína. La co-amplificación de KDR ha sido fuertemente correlacionada con la presencia de amplificación de los genes KIT y/o PDGFFRA en glioblastomas 13, lo que repercute significativamente en la proliferación celular y se ha relacionado con una supervivencia más larga del tumor 14,15.
Tras la caracterización molecular del paciente 2 se identifica una mutación en el gen EGFR, el cual se encuentra localizado en el locus 7p12; dicha posición cromosómica explica que en este caso la mutación se puede deber a una aparición de novo en el tumor más que una segregación en conjunto con los genes de KIT, KDR y PDGFRA11. Es de resaltar que la mutación de EGFR es frecuente en los glioblastomas 16.
El reordenamiento genético en forma de deleción intragénica es el principal mecanismo de mutación oncogénica, en el caso de los gliomas se ha identificado una fusión entre los genes KDR y PDGFRA, que genera una sobreexpresión de estos en los glioblastomas siendo considerada como una fusión de novo por el tumor 17. La sub-representación de la fusión encontrada en estos pacientes podría ser en parte a problemas técnicos, más que a diferencias patogénicas entre los tipos de tumor. El hallazgo en los casos reportados es una inversión paracéntrica con amplificación segmentaria en el cromosoma 4, que da como resultado la fusión del exón 13 del gen KDR con el segmento 10 del gen PDGFRA17; cuando esta mutación está presente, el éxito del tratamiento con inhibidores de PDGFRA se ve disminuido 17, a diferencia de los beneficios que se han encontrado en casos de pacientes que tienen reordenamientos diferentes en estos genes, demostrando así la necesidad de generar un panel molecular que sea específico para la identificación de mutaciones en gliomas, el cual pueda orientar adecuadamente el pronóstico y la identificación de posibles dianas terapéuticas 17.
La radioterapia y la quimioterapia con temozolomida son el tratamiento comúnmente utilizado para los glioblastomas 10; sin embargo, pueden llegar a existir alternativas terapéuticas basadas en las mutaciones somáticas encontradas en el perfil tumoral, ese es el caso de los genes KIT, KDR y PDGFRA, los cuales tienen una frecuencia de amplificación del 4,4%, 3,3% y 8,5% respectivamente 13 Dichas frecuencias pueden variar dependiendo de la población y de la técnica molecular aplicada.
El desarrollo de fármacos anti-tirosina quinasa se ha centrado en encontrar moléculas pequeñas que sean capaces de inhibir la actividad catalítica de las quinasas a través de mecanismos que interfieren con la unión de ATP o sustratos 18. Los inhibidores de RTQ actúa sobre la superficie celular de los receptores que se encuentra normalmente activados en tumores, con lo cual se han desarrollado y aprobado para uso clínico diferentes inhibidores tales como: gefitinib, erlotinib, sunitinib, dasatinib; ya que por medio de ensayos in vivo se ha demostrado que estos reducen la proliferación en líneas celulares de tumores humanos, xenoinjertos y aumentan la apoptosis e inducen la detención del ciclo celular y disminuyen la angiogénesis 19.
Las moléculas pequeñas gefitinib y erlotinib, actúan contra el receptor del factor de crecimiento epidérmico (EGFR) y se han probado en ensayos clínicos para gliomas malignos. En un estudio de fase II de gefitinib para glioblastomas recidivante, se reportó una mediana de supervivencia libre de progresión (mPFS) de 6 semanas 20. En un ensayo de fase I, en el que se usó erlotinib como monoterapia o en combinación con temozolomida, se demostró una respuesta parcial (RP) en el 14 % de los casos y una SLP a 6 meses del 11 % 21.
El fármaco sunitinib es un inhibidor con actividad contra VEGFR-1-3, PDGFR, c-Kit, FLT-3, RET y el receptor CSF-1 (CSF-1R), el cual recibió la aprobación de la FDA para el tratamiento del carcinoma de células renales avanzado y los tumores del estroma gastrointestinal (GIST) 22,23; en el caso de los gliomas, después de demostrar su actividad en estudios preclínicos, ahora se está evaluando en pacientes con glioblastoma, ya sea como monoterapia o en combinación con otros agentes. Las principales toxicidades asociadas con este medicamento fue mielosupresión (neutropenia y trombocitopenia) junto con mucositis 24.
Otro fármaco, usado es el dasatinib con alta especificidad para varias quinasas, incluidas BCR-ABL, Src, c-Kit y PDGFR-b 25, hasta ahora la monoterapia no ha logrado generar cambios en el crecimiento y desarrollo de los gliomas de alto grado, incluso cuando estos fueron seleccionados por la sobreexpresión de PDGFRα 26, pero por sus cualidades intrínsecas como lipófila, tamaño y unión a proteínas, dasatinib es un agente que puede llegar a ser prometedor por su capacidad de penetrar la barrera hematoencefálica; además, su retención está limitada por el líquido cefalorraquídeo (LCR) y la glicoproteína P (P-gp). Trabajos recientes han demostrado que la coadministración de dasatinib con el inhibidor de mTOR everolimus en glioma con mutaciones en PDGFRα aumenta sustancialmente las concentraciones de desatinib lo que ayuda a reducir la progresión tumoral 27.
Si bien no es un inhibidor de RTQ, merece mencionarse el Bevacizumab que es un anticuerpo monoclonal, el cual inhibe la función del factor de crecimiento del endotelio vascular (VEGF) que estimularía el VEGFR, este se caracteriza por ser importante para el proceso de angiogénesis en los tumores, la administración de este medicamento va dirigido a el componente vascular aumentado de los glioblastomas y puede usarse solo o combinado con la quimioterapia generando una reducción significativa del tamaño tumoral, sin embargo, no se ha comprobado que bevacizumab tenga un influencia positiva con el tiempo de sobrevida absoluta. En el año 2009 la Food and Drug Administration (por sus siglas en ingles FDA) aprueba el uso de este fármaco para el tratamiento de los glioblastomas recurrentes 28.
Las terapias dirigidas con inhibidores de los RTQ tienen un gran potencial para el tratamiento de los glioblastomas, sin embargo su eficacia está limitada principalmente por la distribución cerebral deficiente debido a la presencia de la barrera hematoencefálica 29; existen nuevas estrategias farmacológicas como los nanoportadores, ultrasonido enfocado, administración intranasal, sistemas implantables de liberación de fármacos o administración intraarterial, los cuales se están implementado para aumentar las propiedades farmacocinéticas de los inhibidores 29, la adherencia al tratamiento convencional en los pacientes con glioblastoma es menos del 50% (30), esto en gran parte se debe a los cambios genómicos presentados por el tumor, que pueden ir desde mutaciones puntuales hasta metilaciones en las regiones promotoras. De la correcta identificación molecular de dichas variaciones depende si se pueden implementar combinaciones de terapias basadas en el perfil genómico del tumor, un ejemplo de ello son los inhibidores de los RTQ, los cuales aún continúan en estudios clínicos para demostrar el efecto que tiene sobre los glioblastomas, los ensayos que se han realizado hasta el momento no han demostrado resultados significativos, lo cual se puede deber a las debilidades en el diseño y a las técnicas moleculares que se han propuesto para el reconocimiento de las mutaciones en los RTQ, sería importante generar evidencia de la efectividad de los inhibidores para que estos puedan ser utilizados con mayor libertad en la práctica clínica 12,29; todo esto deja como interrogante la necesidad de desarrollar paneles moleculares que sean más exactos y que incluyan genes que funcionen como diana terapéutica.
Conclusión
Se conoce que varias RTQ son activadas en las células tumorales e impulsan el crecimiento y progresión tumoral, la presencia de la amplificación en los genes KIT, KDR y PDGFRA se sugiere como una potencial diana terapéutica, en donde los fármacos anti-tirosina quinasa representan un tratamiento para los pacientes con glioblastoma, siendo probablemente más eficientes que las terapias convencionales, sin embargo, aún falta evidencia clínica que apruebe este tipo de fármacos para el manejo concomitante y/o adyuvante con la temozolomida en este tipo de tumores sólidos.

