











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
La enfermedad de Moyamoya (EMM), entendida como la vasculopatía asociada a la oclusión espontánea de un vaso mayor del polígono de Willis, es una entidad crónica poco común de etiología incierta; su diagnóstico se fundamenta en la presencia de estenosis u oclusión progresiva de las porciones terminales de la arteria carótida interna (ACI) o porción proximal de las arterias cerebral anterior (ACA) y/o cerebral media (ACM), la cual lleva al desarrollo de una red vascular anormal adyacente a los vasos comprometidos, con el fin de compensar la reducción del flujo en la circulación cerebral anterior 1-3. La asociación de esta enfermedad con alguna otra entidad primaria la convierte en un síndrome, entre cuyas causas destacan la neurofibromatosis tipo 1 (NF-1), el síndrome de Down, la enfermedad de Graves, la irradiación terapéutica y la anemia de células falciformes, entre otras 4. Investigar a fondo el síndrome de Moya-moya es importante, ya que tanto la enfermedad como el síndrome comparten un fenotipo de vasculopatía cerebral. Consideramos que, adicionalmente, el síndrome podría ser desencadenado por la colocación de prótesis endovasculares tipo stents derivadores de flujo, secundario al daño mecánico local, el fenómeno inflamatorio de la pared, la proliferación intimal, estenosis de los vasos, fenómenos trombóticos y la disminución del flujo que se podría producir. De manera aparentemente similar a las enfermedades anteriores se produce una respuesta compensatoria con proliferación de vasos anómalos lenticuloestriados y talamoperforantes con una intención de suplir áreas cerebrales con hipoflujo, pero consecuentemente con su anomalía estructural de pared tienen el potencial de desencadenar sangrado o fenómenos isquémicos con las posteriores secuelas. A continuación describimos una serie de casos, en algunos de los cuales el síndrome de Moyamoya ocurrió como respuesta a la implantación de este tipo de stents (Moyamoya iatrogénico).
MATERIALES Y MÉTODOS
Cuatro pacientes con el síndrome de Moyamoya fueron incluidos en esta serie de casos. De ellos, dos tuvieron como causa primaria la implantación de un stent diversor de flujo; una tercera paciente presentaba una neurofibromatosis tipo 1 como enfermedad de base, y en la cuarta paciente no fue posible establecer una causa primaria. En todos los casos el daño crónico ocurrió en la circulación cerebral anterior, bien en la carótida interna o en la cerebral media, o en ambas arterias. Todos los pacientes permanecieron libres de síntomas neurológicos isquémicos o hemorrágicos, a pesar del grave daño arterial detectado. Se detallan a continuación.
Caso clínico 1
Mujer de 39 años de edad con antecedente de neurofibromatosis tipo 1, sin afectación familiar por síndromes neurocutáneos. Acudió a la consulta médica por un síndrome vertiginoso consistente en sensación de giro de los objetos, sin afectación auditiva ni de pares craneanos. Una resonancia magnética (IRM) de cráneo simple y contrastada (figura 1) mostró una hipoplasia del segmento A1 derecho, ausencia de la rama superior de M2 izquierda, sin la presencia de fenómenos isquémicos de dicho hemisferio; el vacío de señal en múltiples sitios en el territorio que debería estar ocupado por la rama superior de M2 sugirió un patrón de Moya-moya, que fue comprobado mediante arteriografía invasiva (figura 2). En esta se confirma el fenómeno de Moyamoya ("bocanada de humo") adyacente a la rama inferior de M2 izquierda; estos vasos neoformados reconstituyen el flujo en el tronco M2 superior ocluido crónicamente. Además hay una disminución del tamaño de los segmentos M1 y M2 de la arteria cerebral media izquierda. En ningún momento la paciente presentó déficit motor o sensitivo, u algún otro signo al examen físico que pudiera ser explicado por los hallazgos angiográficos.
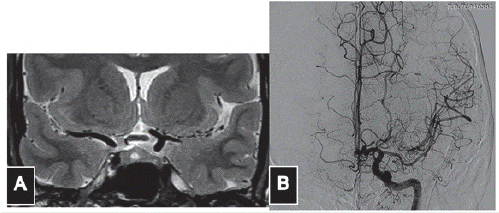
Figura 1 Imagen de resonancia magnética donde se observa una configuración anómala de la porción M1 de la arteria cerebral media izquierda. (A). Angiografía cerebral donde observamos ausencia del tronco superior de M2 luego de la bifurcación (B).

Caso clínico 2
Paciente femenina de 32 años, quien consultó por crisis de cefalea inespecífica que hacía pensar en un patrón migrañoso; sin antecedentes personales importantes, no hipertensa ni fumadora. En una angioresonancia se encontró imagen de aneurisma del sifón carotídeo izquierdo paraoftálmico de 6 mm y cuello ancho. Se llevó a procedimiento endovascular y se le implantó un stent desviador de flujo (figura 2a). El control angiográfico a los 6 meses reveló una estenosis significativa de ACM secundaria a hiperplasia endotelial (figura 2b). Un año después una angiografía mostró un claro patrón de Moyamoya como respuesta a una severa estenosis del segmento M1 de las ACM (figura 2c). La paciente permaneció asintomática en todo momento, libre de molestias que sugirieran fenómenos isquémicos o hemorrágicos.
Caso clínico 3
Paciente femenina, 58 años, hipertensa no fumadora. En 2014, debido a síntomas visuales inespecíficos (visión borrosa ocasional), le practicaron angioRMI en las cuales se apreciaron aneurismas cerebrales múltiples en ambas circulaciones carotídeas. Fue llevada a procedimiento endovascular del eje carotídeo derecho en el cual se implantó un stent desviador de flujo. Una angiografía cerebral 2 años después reveló un severo compromiso de la circulación en el trayecto de la carótida derecha, desde el segmento supraclinoideo hasta el origen de la ACM. Además se aprecia el desarrollo de múltiple neovascularización e hiperplasia lenticuloestriada, conformando un patrón de Moyamoya que preserva la circulación en el territorio de la ACM distal. En el curso de su evolución en dos años la paciente en ningún momento ha presentado compromiso neurológico que sugiera un evento isquémico o hemorrágico.
Caso clínico 4
Paciente femenina con pérdida transitoria de la memoria, sin otros déficits, quien fue internada para estudios, evidenciando en imagen de resonancia magnética infarto antiguo frontal-basal izquierdo, así como una imagen de ausencia de flujo en circulación de la arteria cerebral media izquierda (figura 3 a-b). Holter de 24 horas normal, ecocardiografía transesofágica que reveló un foramen oval permeable, con dopler de miembros inferiores sin lesiones. Doppler carotideo sin alteraciones. La angiografía cerebral invasiva reveló ausencia de flujo en la arteria cerebral media del lado izquierdo con una configuración de Moyamoya compensatoria a expensas de vasos lenticuloestriados que reconstituyen las ramas corticales en ausencia de infartos (figura 3 c-d-e). La paciente ha permanecido libre de síntomas que sugieran isquemia o hemorragia cerebral.

Figura 3 Imágenes de resonancia magnética (A-B-C-D): se evidencia infarto frontal antiguo y ausencia de señal en el territorio de la arteria cerebral media y arteria comunicante anterior del lado izquierdo. Panangiografía a cerebral (E-F-G): reconstitución de la circulación del hemisferio izquierdo a expensas de ramas lenticuloestriadas neoformadas y circulación colateral.
DISCUSIÓN
La enfermedad de Moyamoya se caracteriza por una estenosis y oclusión progresiva de las arterias carótidas internas de ambos lados, así como de sus principales ramas proximales. La estenosis/oclusión vascular está acompañada por el crecimiento de una red de pequeños vasos colaterales lenticuloestriados y talamoperforantes, los cuales tienen una apariencia angiográfica similar al humo que sale después de un puf de cigarrillo (Moyamoya en japonés) 3,4. Esta entidad fue descrita por primera vez en el año 1969 por Suzuki y Takaku 5,6.
Aun cuando su etiología no es muy clara, estudios recientes han logrado establecer una asociación genética con la enfermedad con base en la identificación de alteraciones en los cromosomas 3, 6, 8 y 17. La asociación familiar de esta entidad se ha estimado en un 10 % en Japón y un 6 % en Estados Unidos, y se le ha atribuido a un patrón de herencia autosómica dominante de penetrancia incompleta 7. Se estima entonces un riesgo del 6-12 % de desarrollar la enfermedad si una persona tiene un familiar de primer grado de consanguinidad 4,8,9. Tanto la incidencia como la prevalen-cia de esta enfermedad van en aumento, probablemente por una más frecuente detección, dado el avance en los métodos diagnósticos y el uso de medios no invasivos como la TAC y la IRM. La enfermedad se distribuye alrededor del mundo, sin embargo continúa siendo más alta en los países asiáticos, especialmente en Japón, Corea y China, donde la incidencia es aproximadamente 10 veces la de los países occidentales. Se han establecido dos picos de aparición, alrededor de los 10 años y entre los 30 y los 45 4,10,11.
La manifestación clínica más común, independientemente de la edad de aparición está dada por síntomas isquémicos (accidentes cerebrovasculares isquémicos, isquemia cerebral transitoria, convulsiones), pero las manifestaciones hemorrágicas también cobran importancia especialmente en la edad adulta (donde es siete veces más común que en niños). Otro de los síntomas comunes en esta entidad es la cefalea, la cual característicamente es tipo migrañoso y refractaria al tratamiento médico, y que probablemente se explica por estimulación meníngea por parte de los vasos colaterales dilatados 3,10.
Histopatológicamente la oclusión de los grandes vasos se debe al engrosamiento fibrocelular de la túnica íntima, la ondulación irregular de la lámina elástica interna y el debilitamiento de la túnica media. Por otro lado, característicamente los vasos de Moyamoya que constituyen la circulación colateral presentan depósitos de fibrina en la pared, una lámina elástica característicamente fragmentada, una túnica media debilitada y tienen la predisposición a la formación de microaneurismas, lo cual puede explicar la hemorragia como una de las manifestaciones clínicas de la enfermedad 10.
La enfermedad involucra usualmente ambos hemisferios cerebrales, aunque el compromiso unilateral no hace de un patrón de Moyamoya un síndrome; por el contrario, si no hay presencia de enfermedades subyacentes que expliquen su causa, se rotularía esta como una enfermedad de Moyamoya probable. En niños sucede todo lo contrario, la afectación unilateral puede considerarse como enfermedad y no se enmarcaría como en los adultos como entidad probable 4,5. En la definición anterior de EMM, las características más cruciales de la enfermedad son la localización específica en el polígono, la oclusión vascular, la afectación bilateral y su etiología incierta 12,13.
Los hallazgos angiográficos típicos de la enfermedad de Moyamoya, la estenosis / oclusión de la ACI distal y el desarrollo de colaterales basales no son específicos de la enfermedad. A veces esta vasculopatía se desarrolla en pacientes con otras enfermedades bien caracterizadas, saliendo por ende de la denominación de "enfermedad". Hay muchos tipos de enfermedades asociadas a un patrón vascular de Moyamoya, tales como: la enfermedad arterioesclerótica, enfermedades autoinmunes (lupus eritematoso sistémico, síndrome antifosfolípido, periarteritis nodosa, síndrome de Sjögren), meningitis, neurofibromatosis tipo 1 (NF1), tumores cerebrales, enfermedad de Ghausen, síndrome de Down, trauma encefalocraneano, irradiación cerebral, hipertiroidismo, estenocefalia, síndrome de Turner, síndrome de Alagille, Noonan y Marfan, esclerosis tuberosa, enfermedad de Hirschsprung, enfermedad de depósito del colágeno tipo 1, síndrome de Prader Willi, tumor de Wilms, oxalosis primaria, anemia de células falciformes y de Fanconi, esferocitosis, granuloma eosinofílico, deficiencia de plasminógeno tipo 2, leptospirosis, deficiencia de piruvato kinasa, deficiencia de vitamina C, hiperplasia fibromus-cular, osteogénesis imperfecta, poliquistosis renal, uso de anticonceptivos orales y uso de drogas de abuso (cocaína, etc) 1. Muchas de las afecciones asociadas a Moyamoya son entidades infrecuentes y usualmente sólo una pequeña fracción de los pacientes desarrolla síndrome Moyamoya durante el curso de la enfermedad subyacente.
El síndrome de Moyamoya puede afectar a personas de todas las razas. Los hallazgos angiográficos, así como los síntomas, pueden variar en un amplio rango, desde criterios imaginológicos para enfermedad de Moyamoya (pero con presencia de enfermedad subyacente), hasta aquellos similares a procesos arterioescleróticos, realce y afectación de la vasculatura luego de irradiación, etc.
En uno de los casos que reportamos, la paciente tenía como enfermedad de base una neurofibromatosis tipo 1 y un patrón de Moyamoya en las imágenes realizadas. Creemos que, compartiendo un fenotipo similar pueden tener una patogénesis común en la oclusión vascular y el desarrollo de vasos colaterales. Consideramos que en nuestra paciente, y probablemente en otros reportados en la literatura, dicho patrón vascular se deba a la presencia de un fenómeno isquémico crónico dado por la oclusión de los vasos proximales al polígono de Willis. Esto llevaría al desarrollo de vasos adyacentes colaterales en la fase intermedia y anastomosis transdurales o transóseas entre la carótida interna y externa en fases tardías. Todo esto como un mecanismo compensatorio en un esfuerzo por suplir de oxígeno y nutrientes a un cerebro enormemente dependiente. Las nuevas arterias llamadas vasos de Moyamoya, atraviesan los ganglios basales y el tálamo, suministrando un flujo colateral a las áreas distales hipoperfundidas 6,14. En algún momento de la evolución, el neosistema vascular se hace incompetente y la vasculopatía progresa, tal como lo evidencia la clasificación de Susuki (tabla 1), a tal punto de hacerse sintomática.

Neurofibromatosis tipo 1 y síndrome de Moyamoya.
La enfermedad de Von Recklinghausen o neurofibromatosis tipo 1 es un trastorno autosómico dominante, causado por la pérdida de una línea germinal en un alelo del gen NF1. Lo caracterizan la aparición de manchas café con leche, neurofibromas cutáneos, efélides axilares, nódulos de Lisch, displasias óseas y gliomas del nervio óptico.
Tiene una incidencia de 1 por cada 2500 a 3000 niños 16. La presencia de un síndrome de Moyamoya con NF-1 como entidad desencadenante es rara. Aunque la enfermedad de Moyamoya ocurre frecuentemente en países asiáticos, la mayoría de descripciones con asociación síndrome de Moyamoya-NF1 (SMM-NF1) se han realizado en Europa y Estados Unidos 2. Se ha encontrado que uno de los genes de la enfermedad de Moyamoya familiar se localiza en el cromosoma 17q25, que está en estrecha relación con el gen de la NF1 situado en 17q11.2 17. Esto sugiere la posibilidad de alelismo de estos 2 genes 2,18,19, aunque se han descrito casos con mutaciones de novo. La historia natural de este trastorno es variable. La progresión de la enfermedad puede ser lenta con escasos episodios intercurrentes, o fulminante con un rápido declive neurológico 19. Estudios longitudinales evidencian una afectación contralateral hasta en un 40 % de los pacientes que inicialmente se presentan con afectación unilateral. Algunos estudios indican que el porcentaje de progresión es alto incluso entre los pacientes asintomáticos 19,20, otros lo contrario, con probabilidades relativamente bajas 3,4. Los ictus isquémicos se desarrollan principalmente en niños, mientras los eventos hemorrágicos son más de la población adulta 19. El sangrado puede ser de localización intraventricular, intraparenquimatosa (generalmente a nivel de los ganglios basales) o subaracnoidea, este último atribuido a la rotura de las colaterales frágiles que aparecen conforme progresa la estenosis, o producto de la ruptura aneurismática. Estudios patológicos han puesto de manifiesto que las arterias afectas no muestran cambios ate-roscleróticos ni vasculíticos que produzcan la oclusión 20, sino que esta se produce por la combinación de hiperplasia de células musculares lisas y trombosis intraluminal 3.
NF-1 y vasculopatía cerebral. La neurofibromatosis tipo 1 ocasionalmente causa enfermedad vascular sistémica, con vasculopatía cerebral variable, yendo desde la estenosis/oclusión hasta la dilatación aneurismática, con prevalencias que varían desde 2,5 al 6,4 % 21,22. Algunos muestran estenosis difusa de la ACI en su segmento petroso, estenosis de la ACM distal sin patrón Moyamoya y dilataciones aneurismáticas 21,23,24. El NF1 es un gen supresor de tumores que inhibe la progresión del ciclo celular. Además, induce la producción de neurofibromina, la cual se expresa en células endoteliales vasculares y células de músculo liso. La pérdida de NF1 puede evocar una proliferación inapropiada de células vasculares endoteliales y de células del músculo liso que conduce al engrosamiento y estenosis de la pared vascular 4,25. Aproximadamente el 15 % de los pacientes con NF-1 tienen un glioma del nervio óptico, siendo más frecuente la vasculopatía cerebral en ellos 4,21,23. Además, los pacientes con NF-1 tienen tres veces más riesgo de desarrollar el síndrome de Moyamoya tras la irradiación de los tumores cerebrales, incluidos los gliomas ópticos, en comparación con los pacientes que no reciben el mismo tratamiento 26.
Lesión crónica vascular secundaria al uso de stents desviadores de flujo. Los stents desviadores de flujo, introducidos en el 2007, han ofrecido una nueva alternativa para el tratamiento de los aneurismas cerebrales, especialmente aquellos complejos en razón de su gran tamaño o su cuello ancho, logrando que a través de una malla fina la íntima del vaso afectado se desarrolle de nuevo a lo largo del cuello de la lesión, reconstruyéndolo para excluir al aneurisma de la circulación cerebral, teóricamente sin afectar la circulación colateral 27, a través de la modificación de los desajustes hemodinámicos que propician la formación del saco aneurismático; todo esto, al menos teóricamente, favorece finalmente la trombosis del aneurisma 28. Sin embargo, como cualquier intervención médica, estos dispositivos no están exentos de efectos adversos, y uno de estos, objeto del presente estudio, es la estenosis del vaso en el sitio de la colocación del stent. Es así como la implantación de un stent diversor de flujo fue causa de accidente cerebrovas-cular isquémico en el 2,8 % de los pacientes incluidos en el ensayo Pipeline for Uncoilable or Failed Aneurysms29. Estudios basados en el seguimiento de cohortes de pacientes en quienes se implantó el dispositivo sugieren que este fenómeno podría tratarse de una reacción de la pared del vaso a la presencia del cuerpo extraño, que dicho de paso, carga con una masa considerable de diversas aleaciones metálicas; se ha encontrado que se trata de un fenómeno temprano que ocurre generalmente durante los dos primeros meses post-implantación del diversor 30. El factor de riesgo más importante asociado al desarrollo de la estenosis en el sitio de implantación es la suspensión de la terapia antiplaque-taria, pero es importante tener presente que no ha establecido aún cual es la mejor combinación de medicamentos, ni cuál debería ser la duración exacta de esta terapia tras la intervención para evitar este efecto adverso 31. Diferentes estudios publicados a la fecha, sugieren que la hiperplasia endotelial y la estenosis u oclusión secundaria del vaso que alberga el dispositivo diversor se puede presentar entre un 10 y un 39 % de los pacientes; no obstante es clara la necesidad de estudios adicionales que proporcionen más evidencia al respecto 31.
Consideramos que un stent desviador de flujo entonces, puede producir en algunas ocasiones un daño tal del vaso, que propicie los fenómenos inflamatorios y de estenosis del mismo, con la posterior isquemia silente y compensación secundaria de una red de vasos de Moyamoya como ocurrió en los casos presentados.
Enfoque terapéutico del síndrome de Moyamoya.
El tratamiento farmacológico o quirúrgico no revierte el proceso primario de la enfermedad, pero sí puede prevenir eventos cerebrovasculares y mejorar el flujo sanguíneo cerebral, limitando la neoformación de pequeños y débiles vasos colaterales. Vasodilatadores, antiplaquetarios y anticoagulantes se han usado, aunque su eficacia no ha sido claramente demostrada. En la edad pediátrica, el consenso de expertos de la Asociación Americana del Corazón y Stroke (AHA) recomienda el uso de aspirina (recomendación IIb, nivel de evidencia C). Excepto en pacientes con frecuentes eventos isquémicos a pesar del manejo antiplaquetario y quirúrgico, los anticoagulantes no son recomendados por el riesgo de sangrado y la dificultad de mantener niveles terapéuticos (recomendación clase III, nivel de evidencia C)6,32.
El tratamiento quirúrgico. La cirugía de revascularización es útil en casos seleccionados (recomendación clase I, nivel de evidencia B) 32. Se utilizan dos métodos quirúrgicos: directo e indirecto. En la revascularización directa, una rama de la arteria carótida externa (arteria temporal superficial) se anastomosa directamente a una arteria cortical. La revascularización indirecta es la colocación del tejido vascularizado por ramas de la arteria carótida externa (dura, músculo temporal o la arteria temporal superficial) en contacto directo con el cerebro, para permitir la neoformación de vasos sanguíneos en la corteza cerebral subyacente (encefaloduroarteriosinangiosis, encefalomioarteriosinangiosis, sinangiosis pial y durapexia 6,12,13.
CONCLUSIONES
Tanto la enfermedad como el síndrome de Moyamoya tienen un proceso vasculítico similar de base, que ocasiona la neoformación de una red de vasos que intentan compensar las estenosis de los grandes vasos del polígono de Willis. El patrón de vasos perforantes neoformados debe ser entendido como una respuesta defensiva que protege del daño isquémico distal al vaso principal lesionado.
El síndrome de Moyamoya secundario a la implantación de stents desviadores de flujo, que podría ser clasificado como de origen iatrogénico, hasta donde sabemos no ha sido descrito previamente en la literatura. Faltan aún más estudios que nos indiquen cual es la evolución de los pacientes con stents implantados a largo plazo. Sólo el seguimiento juicioso de ellos dará cuenta de cómo es la evolución del síndrome aquí descrito y cuál será el mejor proceder.

