











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
Digestive functional disorders have been redefined by the Rome Foundation in the Rome IV criteria with a new terminology called the disorders of gut-brain interaction (DGBI)1, and these are the most common diagnoses in gastroenterology. These conditions are highly prevalent and affect up to 40% of people at any given time, and two-thirds of these people will have chronic and fluctuating symptoms, some of them of great intensity1. Given the broad spectrum of therapeutic possibilities with which DGBIs are addressed, the concept of neuromodulation, with drugs acting on the neurogastroenterological dysfunction of these disorders, extending from the central nervous system (CNS) to the enteric nervous system (ENS), has also been renamed by the Rome Foundation2 since their use as psychiatric drugs in order to remove the stigma from them and take advantage of their therapeutic benefits.
This review aims to update and specify the indications for neuromodulators in the main DGBIs, and this first section addresses the reasons for their use.
Rationale
Brain-gut axis, its bases
The gastrointestinal (GI) tract has the vital function of acquiring nutrients through its motor, secretory, and absorptive functions. It constitutes a barrier for the luminal content that protects against potentially pathogenic and antigenic substances or substrates of food or own microbiota3. All of the above requires monitoring events in the intestinal wall and within the intestinal lumen to orchestrate reflexes that elicit appropriate patterns of motility, absorption, secretion, and blood flow to digest and absorb or to dilute and expel GI contents. The ENS and immune system mechanisms play a critical role in activating these reflexes by relaying sensory information to enteric reflex circuits, which provide local control and then inform the CNS to provide an afferent response via afferent pathways3-5.
Enteric nervous system
It is universally known as the intestinal brain; it has a hierarchical neural organization whose objective is to regulate time after time the activity of GI glands, muscles, and circulation in such a way that patterns of secretion and motility are generated during the states and phases of digestion and interprandial periods6,7. The ENS has synaptic connections between sensory neurons, interneurons, and motor neurons. Interneurons are synaptically interconnected in neural networks, which process information about the state of the gut and contain a “library” of programs for different behavior patterns. Motor neurons innervate muscle, secretory glands, and blood vessels. The musculomotor neurons initiate or inhibit the contractile activity of the musculature and determine its force of contraction minute by minute. Secretory motor neurons stimulate the secretory glands to secrete chloride, bicarbonate, and mucus and determine the osmolarity and fluid content of the lumen6,7.
The interaction of the SNE with the interstitial cells of Cajal (ICC) is a determining factor of each motility program stored in their library. ICCs are non-neuronal pacemaker cells that connect to form syncytial electrical networks that extend around the circumference and the long axis of the small and large intestines. ICC networks generate pacemaker electrical potentials (also called slow electrical waves) that spread across gap junctions in circular or longitudinal intestinal muscle, depolarizing the muscle to the threshold action potential and triggering contractions8. Generally, a normal ENS is essential for a healthy gut and the absence of irritating symptoms, such as those associated with the diagnostic criteria for any DGBI. Any neuropathic change in the ENS will most likely result in symptomatic bowel. Functional propulsive motility and its integration with specialized secretory functions cannot work without the ENS7. Pain originating in the GI tract ascends to the brain via the same trineuronal pathways that transmit noxious somatic stimuli (Figure 1).
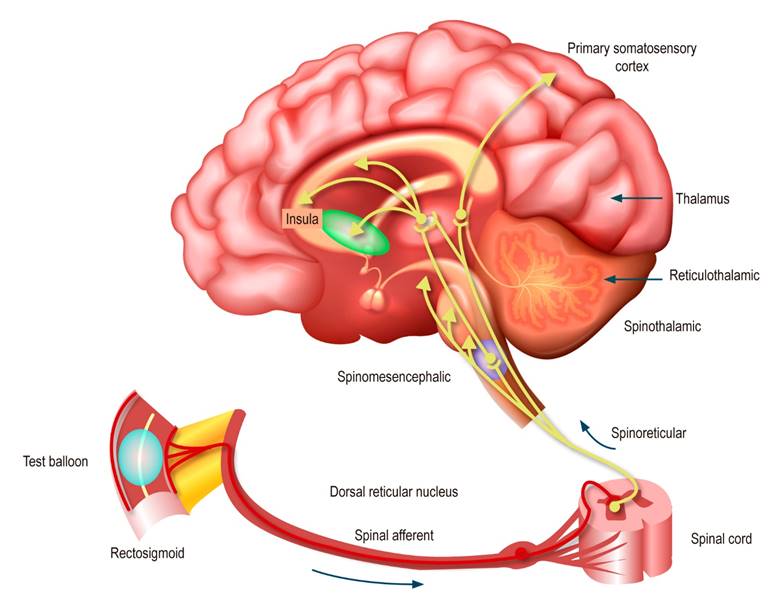
Figure 1 Afferent neural pathways in the DGBI2. Modified with permission from: Drossman DA et al. Gastroenterology. 2018;154(4):1140-1171.e1.
Central processing via hubs and connectomes
Visceral stimuli from the periphery are transmitted to the dorsal horn of the spinal cord, where the first synapse occurs. The last primary synapse occurs on cell bodies within the brain. For the spinothalamic tract, the third-order neuron is within the thalamus, which acts as the main hub for the central pain matrix9. The thalamus is anatomically organized so that noxious signals from the spinal cord are sent to specific regions of the primary somatosensory cortex for signal localization. In contrast, cortical localization of visceral pain is typically less precise since the ascending signal from the spinal cord innervates at multiple levels, and visceral pain signal sources and somatic sources can be transmitted by the spinal cord and second-order neurons (viscerosomatic convergence). Within the central pain matrix, the thalamus sends signals to brain regions that process the emotional component of the pain signal, such as the amygdala, insula, anterior cingulate cortex, hippocampus, and nucleus accumbens. Under normal conditions, activating the central pain matrix provides the appropriate behavioral responses (unpleasant emotion, vigilance, or immobilization of the affected site) to promote recovery and learn to avoid it to prevent future injuries3,9.
Currently, the interconnectivity of pain (visceral and somatic signals) processing nuclei and matrices seeks to be interpreted under the concept of connectomes10, which bring together particular functional and neuroanatomical areas responsible for this function, as verified through multiple neuroimaging11,12.
The afferent response after central processing is directed by descending projections from brainstem nuclei, including the periaqueductal gray, raphe nucleus, locus coeruleus, and lateral rostral ventral medulla down to the dorsal horn of the spinal cord, which modulates the transmission of afferent pain at the level of the first synapse. These descending pathways are controlled top-down by brain regions, particularly the amygdala and perigenual anterior cingulate cortex. Notably, these projections are primarily opioid, noradrenergic, and serotonergic in nature; as noted below, antidepressants can profoundly interfere with these modulatory processes (Figure 2)3.
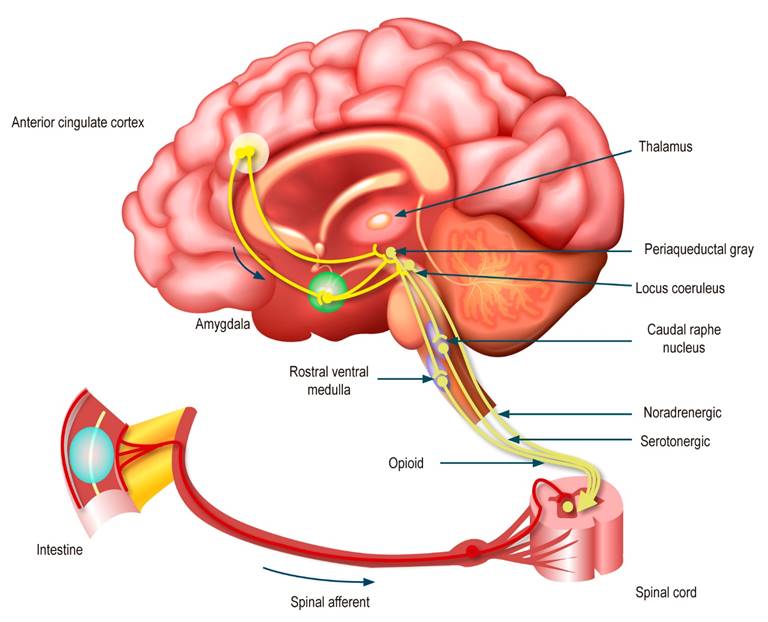
Figure 2 Afferent neural pathways in the DGBI2. Modified with permission from: Drossman DA et al. Gastroenterology. 2018;154(4):1140-1171.e1.
Hypersensitivity: Peripheral and central sensitization
Following an inflammatory event or GI injury, sensory neurons express receptors activated by mediators released from various source cells within the intestinal wall. Neurotrophins, for example, may play a role in axonal conduction and remodeling of sensory innervation after noxa. Both nerve growth factor and glial-derived neurotrophic factor are important in the adaptive response to nerve injury and inflammation13. Many other mediators from blood vessels, muscles, neurons, platelets, leukocytes, lymphocytes, macrophages, mast cells, glia, and fibroblasts are released during inflammation, injury, or ischemia. Some mediators act directly on sensory nerve endings, and others work indirectly, causing the release of other agents from nearby cells. It has been called sensitizing inflammatory soup since it contains amines, purines, prostanoids, proteases, and cytokines, which act on sensory nerve terminals to increase mechanical and chemical sensitivity. This activity as neuromediators, together with the neuropeptides that prevail after acute inflammation, induce phenomena known as ENS neuroplasticity, which would ultimately be responsible for the perpetuation of peripheral hypersensitivity (Figure 3)13.
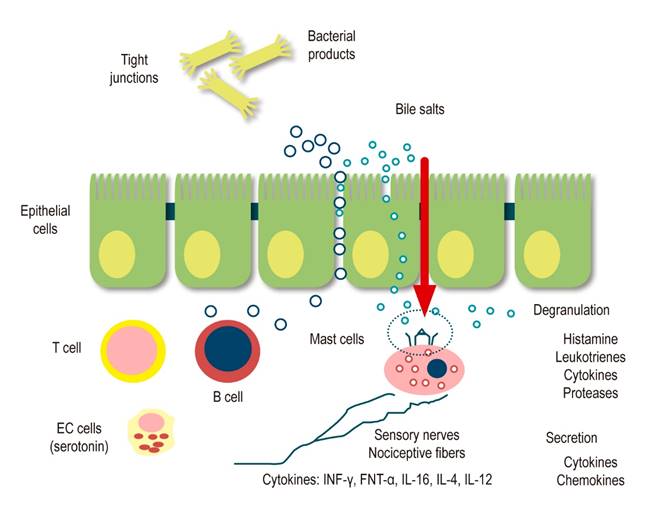
Figure 3 Peripheral sensitization: Intestinal permeability and local inflammatory response15. Modified from: Boyer J et al. Neurogastroenterol Motil. 2018;30(1).
It has also been proposed that barrier dysfunction and intestinal permeability allow the passage of products and antigens from the GI microbiota, produce low-grade inflammation with multiple neuroimmune interactions, and can drive signaling and sensitization with hypersensitivity14. These afferent stimuli trigger reflexes that coordinate bowel function, such that sensitization can also cause hyperreflexia or dysreflexia and thus impaired transit, resulting in diarrhea or constipation, or both. Peripheral sensitization usually develops rapidly and is relatively short-lived. However, in persistent injury or inflammation, sensitization can be prolonged and lead to changes in gene expression. These genes can alter the expression of channels, receptors, or mediators in sensory neurons that modify the amount and pattern of neurotransmitters released by central nerve terminals in the brain and spinal cord. This changes how sensory signals are processed within the CNS, contributes to “central sensitization,” and can prolong hypersensitivity beyond the acute period of injury or inflammation12,13.
Sensory neurons send visceral signals to the ascending spinal pathways via glutamate and neuropeptide tides. These transmitter mechanisms are upregulated in response to inflammation or injury and contribute to hypersensitivity in the brain and spinal cord, where there are central neuroplastic changes called central sensitization, which contribute to chronic pain. Within the dorsal horn of the spinal cord, two mechanisms increase pain signals to the brain: increased synaptic transmission via glutamate, calcitonin gene-related peptide, and substance P in ascending excitatory pathways or decreased descending inhibitory modulation3.
In the brain, sensitization can occur in second-order spinal neurons, such as the thalamus, periaqueductal gray (PAG), parabrachial nucleus, and locus coeruleus. Increased signaling from those nuclei promotes neuroplasticity and abnormal pain processing within the extended pain matrix (prefrontal cortex [PFC], anterior cingulate cortex, amygdala, insula), amplifying discomfort and negative emotions associated with chronic visceral pain or a decrease in the descending pain inhibitory system through the PAG and rostral ventral medulla4. Notably, the amygdala is a critical nucleus that integrates noxious visceral signals with anxiety/fear behaviors, and its hyperactivation could influence multiple central pain matrix nuclei and descending brainstem nuclei that modulate the amygdala function. Numerous neuroimaging studies have also shown functional, connectivity, and structure differences between irritable bowel syndrome (IBS) patients and healthy controls. Thus, central sensitization may promote chronic abdominal pain in IBS through the remodeling of connections both within the brain and spinal cord16,17.
The brain-gut axis is the bidirectional neurohumoral communication system between the brain and the gut, which continuously sends signals about homeostatic information and the physiological condition of the digestive system to the brain through afferent (spinal and vagal) and humoral nerves of the ENS or “cerebral gut.” Under normal physiological conditions, most of these gut signals are not perceived consciously. However, the subjective experience of visceral pain results from the conscious perception of directed gut-brain signals induced by noxious stimuli. It indicates a potential threat to homeostasis, requiring a behavioral response18.
Visceral pain perception does not show a linear pattern but rather arises from a complex psychobiological process whereby the intensity of peripheral afferent input is processed and continuously modulated by cognitive and affective circuits in the brain and through descending modulators. These mechanisms help to understand the influence of cognitive and affective processes on the perception of GI symptoms in patients with DGBI and the therapeutic effect of interventions directed at these processes that constitute the basis for understanding DGBI as a model of gut-brain signaling disorders. In addition, dysfunction of these modulatory systems could allow non-noxious physiological stimuli to be perceived as painful or unpleasant (visceral hypersensitivity), leading to chronic visceral pain or discomfort, a hallmark of DGBI symptoms18.
Aspects of a biopsychosocial model
The rationale for using neuromodulators has an even broader context when the factors contributing to developing functional gastrointestinal disorders (FGID)-DGBI are appreciated. Here, we highlight some of the most relevant: although there is evidence of the contribution of genetic, epigenetic, and cultural components, stressful life events (including sexual abuse, physical abuse, and emotional abuse, particularly during childhood), compared with controls. Patients with IBS report a higher prevalence of this type of adverse event, and, generally, this history is related to greater severity of FGID with worse outcomes, clinical stress, and impairment of daily functioning and quality of life (Figure 4)18,19.
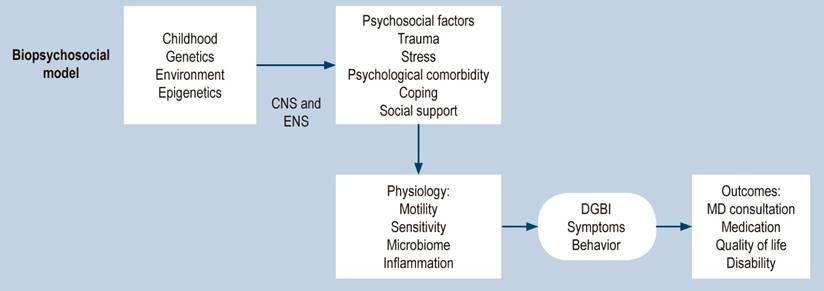
Figure 4 Biopsychosocial model for DGBI2. Modified with permission from: Drossman DA et al. Gastroenterology. 2018;154(4):1140-1171.e1.
Affective disorders
The overlap between depression and DGBI is around 30% in primary care settings and slightly higher in tertiary care. Depression can affect the number of functional GI symptoms experienced or DGBI diagnoses. Suicidal ideation is present between 15% and 38% of patients with IBS, and it has been related to hopelessness associated with the perception of the severity of the symptoms, the impact on the quality of life, and the variable and sometimes poor response to treatment. Likewise, the comorbidity of depression has been related to poor outcomes, increased use of medical services and cost of medical care, functional impairment, poor quality of life, and poor commitment to treatment outcomes20.
Anxiety disorders
They are the most common psychiatric comorbidity in 30%-50% of patients with DGBIs. They can initiate or perpetuate symptoms through their association with arousal in response to stress and alter GI sensitivity and motor function, as well as sensitivity to anxiety itself, hypervigilance of the body, and the ability to tolerate discomfort and pain20.
Somatic symptom disorder
The Diagnostic Manual of Mental Disorders, 5th Edition, modified the concept of somatization in favor of somatic symptom disorders. In the new diagnostic category, somatic symptoms may or may not have a medical explanation but are distressing and disabling and are associated with excessive and disproportionate thoughts, feelings, and behaviors for more than six months. This highly controversial approach shifts the experience of medically inexplicable symptoms as (unconscious) manifestations of psychological disorders towards abnormal cognitive and affective processes (e.g., excessive concern about the disease, concern about the body, and hypochondriasis) as contributors and consequences of symptoms21.
Somatization is associated with GI sensorimotor processes, particularly pain, including gastric tenderness and emptying, symptom severity, and impaired quality of life in functional dyspepsia and IBS. It is also associated with the frequency of use of medical care and predicts a poor response or adherence to treatment. Therefore, assessing somatization by testing the severity of multiple somatic symptoms remains clinically useful. Somatization explains the frequent extraintestinal symptoms of IBS and the high co-occurrence between DGBI and other somatic symptom disorders (SSD), including interstitial cystitis, pelvic pain, headaches, and fibromyalgia18.
Cognitive-affective processes
Some psychological processes may be superimposed; for example, when they include health anxiety (mainly GI), specific anxiety about other symptoms, attentional bias such as symptom hypervigilance, and catastrophizing have been linked to DGBI regardless of psychiatric comorbidity, and are essential treatment targets for cognitive-behavioral therapy (CBT)22.

