











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
Porphyrias are disorders of heme synthesis involving either inherited or acquired enzyme deficiencies, resulting in an overproduction of heme precursors1,2. Heme is an essential molecule that performs a wide range of functions necessary for aerobic life as a cofactor in hemoproteins3,4.
Porphyrias are classified into acute hepatic porphyrias (AHP) and photosensitive porphyrias. AHPs result from hepatic overproduction of porphyrin precursors, aminolevulinic acid (ALA), and porphobilinogen (PBG), and include acute intermittent porphyria (AIP), variegate porphyria (VP), hereditary coproporphyria (HCP), and 5-aminolevulinate dehydratase deficiency porphyria (ALAD-P). Clinical manifestations are characterized by acute neurovisceral symptoms, such as abdominal pain, nausea, vomiting, constipation, muscle weakness, neuropathy, tachycardia, and hypertension5. Symptomatic AHPs affect approximately 1 in 100,000 patients; however, it is estimated that there is a diagnostic delay of more than 15 years from initial presentation5.
ALAD deficiency is a rare autosomal recessive disorder (with only eight documented cases worldwide) in which enzyme activity is less than 3% of normal6,7, resulting in little or no elevation of PBG but marked increases in urinary ALA and coproporphyrin III7. No cases of this type of porphyria have been documented in Colombia.
The initial approach for an acute porphyria episode is a multidisciplinary management strategy ranging from hospitalization to intensive care, depending on the severity of presentation. Key management principles include avoidance of triggering factors, caloric supplementation, and often hematotherapy5,8, with liver transplantation as a last resort8. Long-term complications include liver disease (cirrhosis and hepatocellular carcinoma), hypertension, and chronic kidney disease.
This case report presents a clinical phenotype of ALAD deficiency porphyria with a history of urine discoloration and recurrent abdominal pain. The initial diagnostic approach was diverted due to a suspected diagnosis of paroxysmal nocturnal hemoglobinuria, further complicated by cutaneous manifestations and interstitial nephritis likely associated with the patient’s intake of nonsteroidal anti-inflammatory drugs (NSAIDs) for symptom management. Mutation confirmation was not possible due to discontinuation of follow-up by the patient, but this case contributes to the literature by providing an approach to diagnostic testing and a review of relevant studies.
Case Description
This case involves a 24-year-old male with no significant medical, toxicological, family, or perinatal history. He presented with a four-day clinical course characterized by severe epigastric and bilateral lumbar pain, described as stabbing and rated 10/10 on the visual analog scale (VAS). These symptoms were associated with changes in urine color (Figure 1), pruritic skin lesions, loose stools, emesis episodes, and a documented fever of 38.7°C.

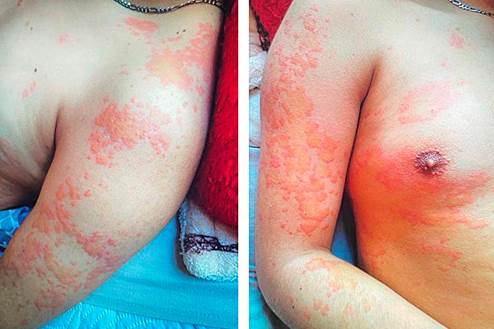
Initial evaluation recorded normal vital signs, diffuse abdominal tenderness on palpation without masses, organomegaly, or peritoneal irritation. Erythematous-edematous pruritic plaques with central pallor were observed, primarily affecting the upper limbs and trunk (Figure 2).
Source: Author’s File.
Figure 2 Pruritic skin lesions presenting as erythematous-edematous plaques with central pallor, predominantly on the upper limbs and trunk.
Initial laboratory findings showed elevated nitrogenous waste, consistent with acute kidney injury (AKI) classified as KDIGO stage III, a slight increase in lactate dehydrogenase (Table 1), and a diffuse increase in liver echogenicity on abdominal ultrasound.
Table 1 Laboratory Report
| Laboratory Tests | Patient Values | Reference Values | Unit of Measure |
|---|---|---|---|
| Lactate dehydrogenase | 510.31 | 120-246 | U/L |
| Chloride | 102 | 98-107 | mmol/L |
| Sodium | 137 | 136-145 | mmol/L |
| Potassium | 4.32 | 3.5-5.1 | mmol/L |
| Prothrombin time | 10.50 | 9.9-11.8 | s |
| INR | 0.98 | 0.90-1.15 | mg/dL |
| Partial thromboplastin time | 28.7 | 25.0-31.3 | s |
| Blood urea nitrogen | 55.52 | 9-23 | mg/dL |
| Alkaline phosphatase | 94.84 | 46-116 | U/L |
| Glucose | 71 | 74-106 | mg/dL |
| Total bilirubin | 0.61 | 0.3-1.2 | mg/dL |
| Direct bilirubin | 0.24 | 0-10 | mg/dL |
| Indirect bilirubin | 0.37 | mg/dL | |
| Glutamic-oxalacetic transaminase | 27.95 | <34 | |
| Creatinine | 11.24 | 0.6-1.1 | |
| Haptoglobin | 138.9 | 40-280 | |
| Parathyroid hormone | 200.1 | 18.5-88 | |
| Antinuclear antibodies | Negative | ||
| Anti-DNA antibodies | Negative | ||
| Hepatitis B | <0.10 | 0-50 | |
| Hepatitis C antibody | 0.08 | 0-1.0 | |
| HIV 1/2 antibodies | Negative | ||
| Cardiolipin IgM antibodies | 10.543 | <20 | |
| Cardiolipin IgG antibodies | 1.927 | <20 | |
| β2 glycoprotein IgM | 6.537 | 0-20 | |
| β2 Glycoprotein IgG | 1.927 | 0-20 | |
| Myeloperoxidase | 1.6 | <20 | |
| δ-aminolevulinic acid | 14.1 | 1-7 |
DNA: deoxyribonucleic acid; IgG: immunoglobulin G; IgM: immunoglobulin M; INR: international normalized ratio; HIV: human immunodeficiency virus. Author’s own research.
Subsequently, during a follow-up interview, the patient reported outpatient management for recurrent abdominal pain occurring every three months, for which he had self-medicated with N-butylscopolamine bromide and diclofenac without clinical improvement. His recent diclofenac use had increased over the past four days, with an approximate daily dose of 300-400 mg. Outpatient studies included abdominal imaging, which showed no abnormalities, normal PBG levels, a negative autoimmune profile, and negative infectious disease testing, including for human immunodeficiency virus (HIV), hepatotropic viruses, and rapid plasma reagin (RPR). Additionally, electrophoresis and immunofixation were negative, although flow cytometry detected paroxysmal nocturnal hemoglobinuria (PNH) clones in monocytes (12.9%), granulocytes (12.5%), and erythroid cells (1.2%). Given the abdominal pain, acute kidney injury (AKI), and urine discoloration, an episode of PNH with secondary abdominal-renal thrombotic events was initially considered as a diagnostic impression. However, this was ruled out due to the low clinical significance of the small PNH clone populations and the absence of a clinical phenotype consistent with PNH.
Due to persistent renal impairment without indications for urgent dialysis, but with reduced urine density and polyuria, a renal biopsy was performed. Histopathology revealed tubulointerstitial nephritis without glomerular involvement or chronic changes. The patient experienced a stationary clinical course with persistent symptoms of limiting visceral pain, associated with somnolence and neuropathy manifesting as dysesthesias in the extremities and tongue. Given the intermittent and temporally related nature of symptoms, as well as a reported association with physical and emotional stress and alcohol intake, additional inpatient studies were conducted. The reports showed elevated urinary ALAD levels, normal porphobilinogen, and elevated 24-hour urinary porphyrins. This clinical presentation was considered compatible with a probable phenotype of ALAD porphyria, leading to treatment initiation with dextrose solution and subsequent administration of human hemin at 4 mg/kg/day (260 mg) via infusion for three days. Improvement was observed in both the reported symptoms and follow-up laboratory values, including a gradual reduction in nitrogenous waste.
The skin manifestations were interpreted as likely related to NSAID-induced skin toxicity or a gastrointestinal infectious agent, and were treated with ampicillin-sulbactam for five days. However, due to spontaneous resolution within 24 hours of admission, a skin biopsy was not pursued. The patient did not continue follow-up, and due to administrative constraints, the mutation panel needed to confirm the type of porphyria could not be completed. Consequently, the case was managed as a probable ALAD porphyria phenotype with initial diagnostic distractions, no PNH phenotype, and complications from NSAID overuse.
Discussion
Acute hepatic porphyrias (AHP) are rare, with estimated prevalence rates of five per 100,000 people3. Among these, ALAD deficiency is the rarest form, with only eight cases documented in the literature, and is the least understood type of porphyria8, with no cases reported in Colombia. The ALAD gene is located on chromosome 9q34 and catalyzes the condensation of two ALA molecules to form PBG8. Its mutation, an autosomal recessive disorder, results in enzyme activity of less than 10%. All individuals affected by this mutation experience severe disease onset early in life3, with all reported cases being male, as is true for this patient, who has had intermittent symptoms since the age of 13.
Clinically, AHPs manifest as intermittent, progressive, colicky abdominal pain radiating to the lumbar region and extremities, along with nausea, vomiting, urine discoloration, and constipation5,6). However, diarrhea can sometimes occur8, as in this case. ALA, PBG, and other hepatic heme precursors can cross the blood-brain barrier, exerting agonistic and antagonistic effects on gamma-aminobutyric acid (GABA) receptors in the central and peripheral nervous systems, leading to delirium, confusion, neuropathies, muscle weakness, seizures, tetraparesis, and respiratory arrest3,5. Physical examination may reveal tachycardia and hypertension due to sympathetic hyperactivity during acute attacks6,7. These episodes may also be associated with hyponatremia and hypomagnesemia, attributed to a combination of hypovolemia and syndrome of inappropriate antidiuretic hormone secretion. Given its incomplete penetrance, triggering factors-such as excessive alcohol intake, smoking, drug use, fasting, or starvation-are necessary and likely act directly to increase hepatic ALA synthase-1 (ALAS1) mRNA levels, which is rate-limiting in heme production, thereby creating a vicious cycle3.
Renal disease associated with porphyria can occur in approximately 59% of patients, with an annual decline in glomerular filtration rate of around 1 mL/min per 1.73 m². Literature suggests that causes may include endoplasmic reticulum stress mediated by ALA and PBG, apoptosis, and phenotypic changes in proximal tubular epithelial cells leading to renal injury, as well as the action of a variant of the human peptide transporter 2 expressed by proximal tubular cells that mediates ALA reabsorption3. Due to the deterioration in renal function, a renal biopsy was performed on this patient, which reported tubulointerstitial nephritis, considered multifactorial: the effect of porphyria combined primarily with chronic NSAID use, with an increase in consumption prior to admission. Liver enzymes may be elevated in 13% of patients during acute attacks, and while cases of cirrhosis have been reported, the cause of elevated liver enzymes and hepatic fibrosis remains poorly understood; thus, abnormal liver enzymes should prompt diagnostic evaluation for alternative etiologies6.
The diagnosis of symptomatic AHP requires biochemical testing, specifically showing significantly elevated ALA and PBG levels in urine or plasma, except in ALAD porphyria, where PBG levels do not rise significantly5,8, as was the case with this patient. It is essential to note that, as precursors of porphyrins, ALA and PBG are not included in standard porphyrin tests. Once biochemical testing indicates ALAD porphyria, confirmation can be achieved through genetic testing with gene sequencing; however, it is not recommended for initial screening6 as it is unnecessary for diagnosis9. Genetic testing is, however, valuable for family studies and identifying previously unknown mutations9. In this patient’s case, diagnostic challenges included a history of paroxysmal nocturnal hemoglobinuria (PNH), which initially led to distraction from considering ALAD porphyria. Although flow cytometry showed positive PNH clones, these were low-quality and lacked a clinical phenotype of the disease, making PNH diagnosis less probable. Additionally, the skin manifestations, initially interpreted as potential NSAID toxicity, further distracted from an early consideration of ALAD porphyria.
Acute management of AHP involves strict avoidance of porphyrinogenic drugs and triggering factors (such as acute illnesses or infections, physical or psychological stress, excessive alcohol intake, and smoking), intensive care monitoring, caloric support (300 grams of glucose per day), effective pain management, electrolyte correction, and often hematotherapy5,6. With early intravenous heme administration, patients begin to improve within 48 hours due to decreased ALAS1 expression4,5. The acute phase may involve hyponatremia and hypomagnesemia, attributed to a combination of hypovolemia and syndrome of inappropriate antidiuretic hormone secretion. For seizures associated with AHP, benzodiazepines are considered relatively safe, and levetiracetam is regarded as a safe option6. In complex cases, therapeutic options include givosiran, a small interfering RNA that neutralizes excess ALAS1 mRNA in hepatocytes, with liver transplant as a last-resort option5.
Differential diagnoses should exclude other AHPs (via PBG measurement), lead poisoning, and hereditary tyrosinemia type 1. In this case, lead poisoning was ruled out due to the absence of anemia, basophilic stippling on peripheral smear, and a “lead line” (bluish pigmentation at the gum line and teeth), along with a detailed history ruling out exposure to lead5.
AHPs are now considered chronic conditions with a significant negative impact on physical and emotional health, resulting in a reduced quality of life4. They pose an increased risk of long-term complications, including hepatocellular carcinoma (HCC) and chronic kidney disease6, requiring annual screening for these complications.
Conclusions
It is essential to recognize the clinical phenotype to accurately interpret porphyria tests; even if initial tests are negative, further evaluation should be pursued to consider a potential phenotype, such as ALAD porphyria. The impact of neurovisceral manifestations often leads to the concomitant use of analgesic medications that may cause toxicity and should always be considered in patients with these conditions. Despite their relative rarity and complexity, most porphyrias can be readily defined and diagnosed. One of the most common causes of diagnostic delay is failing to consider this entity promptly in differential diagnoses. Much remains to be learned about ALAD porphyria.

