











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
La enfermedad de hígado graso no alcohólico (EHGNA) se define como una esteatosis hepática simple a partir del 5 % o más de los hepatocitos1, con un espectro amplio de progresión de esteatosis simple a esteatohepatitis; este proceso se caracteriza por inflamación y fibrosis, que eventualmente puede llegar a ser irreversible, como sucede en la cirrosis y el carcinoma hepatocelular. La definición además debe excluir causas secundarias de acumulación de grasa y lesión en el hígado derivadas del consumo crónico de alcohol, el uso de medicamentos esteatogénicos, hepatitis de origen viral, entre otras enfermedades de carácter agudo como crónico1,2.
La prevalencia a nivel mundial de EHGNA es del 25,24 % para la edad adulta, significativa en todos los continentes1, de los cuales la prevalencia para la población pediátrica general se estima en 7,6 % a 34,2 %3,4. En América Latina, la población está fuertemente asociada a factores de riesgo para esta enfermedad, como sobrepeso, obesidad, resistencia a la insulina, entre otros, lo cual lleva a una prevalencia estimada del 44,37 % en la población adulta1. En un estudio realizado se evaluó la prevalencia de EHGNA en 833 niños con sobrepeso y obesidad de una escuela primaria en México, con edades entre 5,5 y 12 años; se reportó en 12,6 %5. El riesgo de padecer esta enfermedad aumenta conforme la edad, con una distribución variable por sexos en la edad adulta, y es más común en hombres que en mujeres en edad pediátrica4,6.
La EHGNA en el adulto, con base en la teoría de golpes múltiples7, tiene inicio con el primer golpe, representado por la resistencia a la insulina, lo cual ocasiona en los hepatocitos un marcado aumento de la ruta metabólica de lipogénesis de novo hepática y un aumento de la lipolisis por parte del tejido adiposo. Posteriormente, se establece la secreción de adipocinas y citocinas inflamatorias en los adipocitos, mientras que la sobrecarga en la síntesis de novo de lípidos en los hepatocitos promueve el desarrollo de un ambiente lipotóxico, el cual genera disfunción del retículo endoplasmático y un aumento de especies reactivas de oxígeno en la mitocondria, que eventualmente produce estrés oxidante y lesión hepática7. Adicionalmente, la alteración del eje intestino-hígado permite obtener un aumento de ácidos grasos de cadena corta que son transportados al hígado. (ver Figura 1)7,8,9.
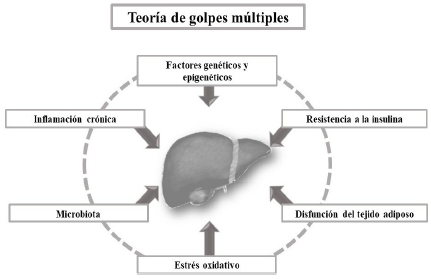
Fuente: tomado y editado de Buzzetti E, et al.7, Kobyliak N, et al.8, y Rahmanabadi A, et al.9.
Figura 1 La teoría de los golpes múltiples. Diferentes entidades patológicas representan cada uno de los golpes de la teoría del golpe múltiple: resistencia a la insulina, disfunción del tejido adiposo, estrés del retículo endoplasmático y mitocondrial, microbiota intestinal, inflamación crónica, modificación del código genético y factores epigenéticos. Cada golpe contribuye al inicio y la progresión de la EHGNA.
Durante el establecimiento de EHGNA y durante toda la progresión de la enfermedad existe una importante capacidad de regulación tanto a nivel genético como epigenético7,10. Relaciones puntuales reportadas en relación con la EHGNA por parte de la genética se han realizado mediante la identificación de polimorfismos de un solo nucleótido, que se distinguen como el cambio de un solo nucleótido en la secuencia genética. Por parte de la epigenética, se han identificado una vasta cantidad de mecanismos epigenéticos, que no modifican la secuencia del ácido desoxirribonucleico (ADN), y que podemos dividir en modificaciones químicas covalentes del ADN mediante la agregación de un grupo metilo al nucleótido citosina, denominado como procesos de metilación diferencial del ADN, que suele representar una disminución de la expresión genética subyacente, y otros dos mecanismos, que son las modificaciones postraduccionales de histonas (PTM) y los diversos tipos de ácido ribonucleico (ARNs), que cuentan con una cantidad extensa y compleja de mecanismos de acción7,10,11.
La EHGNA pediátrica se encuentra fuertemente relacionada con los mecanismos epigenéticos, ya que estos permiten que el feto se desarrolle y adapte ante los diversos tipos de eventos adversos fetales, como diabetes y obesidad gestacional, entre otras alteraciones metabólicas, lo cual le puede predisponer eventualmente al desarrollo de EHGNA en etapas tempranas y posteriores de la vida4,11, por lo que para la edad pediátrica el verdadero "primer golpe" es representado por la regulación epigenética que programa al feto a través de los distintos mecanismos epigenéticos como la metilación diferencial de ADN, para una predisposición al desarrollo de EHGNA, entre otras enfermedades metabólicas como obesidad y diabetes12-17.
El diagnóstico estándar de oro de EHGNA es la biopsia hepática, la cual muestra en pacientes pediátricos como forma más temprana un depósito de grasa e infiltración inflamatoria en zonas periportales, en comparación con una distribución perivenular clásica en los adultos18. Este procedimiento aumenta la comorbilidad y la mortalidad en los pacientes19, por lo que el cribado se realiza mediante biomarcadores séricos como la alanina aminotransferasa y aspartato aminotransferasa. Además, los métodos alternativos de detección y monitorización son estudios de imagen como ultrasonido hepático, elastografía y resonancia magnética para la detección y monitorización20,21.
Por otra parte, la obesidad en el embarazo ha aumentado significativamente durante la actual pandemia de obesidad, que según informó la Organización Mundial de la Salud (OMS) se triplicó desde 1975. Hasta el año 2016, 1900 millones de adultos mayores de 18 años tienen sobrepeso, más de 650 millones son obesos, y de estos, un 40 % de las mujeres tienen sobrepeso y un 15 % son obesas22.
Según las directrices internacionales, el diagnóstico de obesidad en el embarazo se define como un índice de masa corporal (IMC) de 30 kg/m2 o más, utilizando la altura y el peso antes del embarazo o los medidos en la primera visita prenatal si no se dispone de información previa al embarazo23.
El objetivo de la presente investigación fue realizar una revisión sobre la predisposición de EHGNA pediátrica relacionada con la exposición del feto ante la obesidad gestacional, a través de los procesos de metilación diferencial reportados. El estudio de esta enfermedad ayuda en el entendimiento de la fisiopatología, la cual aún no ha sido comprendida en su totalidad. Actualmente, los métodos para la determinación de severidad son limitados y no se cuenta con un fármaco específico. La justificación de esta revisión se fundamenta en la relevancia de la metilación diferencial de ADN en el alcance de un mayor entendimiento sobre los cambios secundarios a la exposición fetal ante obesidad gestacional.
Metodología de búsqueda
Se realizó una búsqueda en la base de datos PubMed desde el primero de enero del año 2021 hasta el 28 de agosto del 2023, con artículos de no más de 5 años de antigüedad desde el 2021, con la excepción de artículos a los cuales se les atribuye el crédito de resultados puntuales y/o conceptos relevantes para la presente investigación, con los términos MeSH individualmente, y en las combinaciones de operadores booleanos a continuación: (offspring) AND (obesity), (nafld) AND (children), (obesity) AND (methylation) y (obesity) AND (fetal programming). Se utilizaron los siguientes criterios de inclusión: ensayos clínicos aleatorizados, revisiones sistemáticas y revisiones de tema en idioma inglés relacionados con los procesos de metilación de ADN que predisponen a EHGNA en edades tempranas a través de exposición fetal a obesidad materna. Se evaluaron inicialmente los títulos y resúmenes de los artículos de acuerdo con los criterios de inclusión, en una primera selección se incluyeron 899 artículos.
Posteriormente, se excluyeron todos los artículos que no tuvieran como tema central los procesos de metilación de EHGNA en la edad pediátrica. Una vez revisados los artículos completos, se obtuvo un total de 376 artículos de los cuales, en una revisión final, fueron excluidos 305, dejando un total de 71 artículos que cumplieron con todos los criterios de selección y que además reportaron resultados significativos sobre la relación de los procesos de metilación diferencial y la exposición fetal a obesidad gestacional para el desarrollo de la presente revisión de tema.
Metilación de ADN en la programación fetal
La hipótesis de los orígenes del desarrollo de la salud y la enfermedad (DOHaD por sus siglas en inglés Developmental Origins of Health and Disease) establece que las adversidades del entorno intrauterino influyen en la estructura y función de las células, tejidos y órganos en desarrollo, lo que genera diferencias fenotípicas entre individuos a lo largo de su vida; esto conceptualiza la preparación de la descendencia para las condiciones previstas al nacimiento a través de alteraciones en la señalización placentaria y programación epigenética24,25.
Este hecho fue descrito por primera vez en estudios humanos mediante la evidencia epidemiológica que mostró asociaciones entre eventos adversos del desarrollo in utero, con el subsecuente riesgo de desarrollar diversas patologías desde la adolescencia hasta la edad adulta25.
La programación fetal intercede cuando el entorno óptimo en el que el feto crece se ve afectado por exposiciones adversas, especialmente durante períodos críticos de desarrollo de órganos esenciales. Esto permite programar al nuevo organismo para mantener la homeostasis en condiciones inadecuadas a través de cambios del fenotipo que determina el comienzo de futuros problemas de salud18,19. Debido a esto la herencia de la genética mendeliana, basada en secuencias de ADN, no sustenta completamente la presencia de enfermedades complejas como el EHGNA en edad temprana26.
Los procesos exactos de cómo funciona la programación fetal aún no han sido totalmente comprendidos, aunque se han confirmado efectos en la descendencia mediante la regulación de los mecanismos epigenéticos, los cuales incluyen la metilación del ADN, modificación de histonas y ARNs no codificantes13, que programan los perfiles de expresión diversificados de todas las células del cuerpo humano sin modificación de la secuencia primaria del ADN27.
Estos mecanismos son transmitidos a la descendencia con base en la herencia intergeneracional, que avanza a lo largo de únicamente dos generaciones, y la herencia transgeneracional, que abarca más de dos generaciones28-30.
Entre los mecanismos epigenéticos se encuentra la metilación del ADN, que modula la expresión de los genes ejerciendo una función dependiente de la secuencia genética subyacente31-33, mediante una modificación covalente en el quinto carbono de la pirimidina, citosina, catalizada por ADN metiltransferasas (DNMT), la cual agrega un grupo metilo transferido por parte de S-adenosilmetionina (SAM) para formar 5-metilcitosina, la principal forma de modificación del ADN34.
Las DNMTs se clasifican con base en la regulación de los procesos de metilación, principalmente en dos categorías: 1) DNMT1 se localiza en la horquilla de replicación donde se forma el ADN hemimetilado recién sintetizado, para funcionar durante la replicación del ADN para copiar el patrón de metilación de la cadena de ADN paterna a la recién sintetizada hebra de ADN hija, y 2) Las DNMT3a y DNMT3b establecen un nuevo patrón de metilación para ADN no modificado en una función de metilación de novo35,36.
La metilación del ADN puede reducir por sí misma la expresión genética mediante una alteración en la unión de los activadores transcripcionales, además de una segunda vía de modulación por parte de una clase de proteínas con una alta afinidad por 5-metilcitosina, que posteriormente también son capaces de inhibir la unión del factor de transcripción37.
La metilación del ADN se produce predominantemente en las islas CpG, las cuales son segmentos de ADN ricos en nucleótidos de citosina y guanina, con alrededor de 1000 pares de bases de longitud. Estas secuencias contienen aproximadamente del 70 al 75 % de todas las regiones promotoras de los genes, y no suelen estar metiladas. Las regiones promotoras restantes se encuentran con densidades de CpG muy bajas, donde la metilación es muy poco probable que regule su expresión38.
La metilación de las islas CpG representa una interferencia con la unión del factor de transcripción y el reclutamiento de proteínas de unión a metilo represivas, silenciando de manera estable la expresión génica39.
La desmetilación del ADN puede ocurrir por dos mecanismos. El primero es la desmetilación pasiva, que ocurre cuando los elementos funcionales que mantienen la metilación del ADN se encuentran en cantidades precarias, lo que puede ocurrir durante la replicación. El segundo es la desmetilación activa, que se lleva a cabo por dos vías principales: 1) convirtiendo 5-metilcitosina a tiamina y posteriormente a citosina mediante las enzimas AID/ APOBEC y TDG, respectivamente; 2) a través de las enzimas TET1, TET2 y TET3, que catalizan la oxidación de 5-metilcitosina en una citosina no modificada40-43.
Evidencia de que la metilación diferencial a nivel sanguíneo produce cambios profundos en el desarrollo fisiológico de los órganos podría encontrarse plasmada en los estudios basados en la población expuesta prenatalmente a la hambruna del invierno holandés, entre los años 1944-1945, donde se presentaron niveles de metilación alterados 6 décadas después de su exposición intrauterina a la desnutrición, en comparación con sus hermanos del mismo sexo no expuestos43,44.
Exposición fetal a la obesidad en los orígenes del desarrollo de EHGNA y su progresión
La intolerancia a la glucosa y el aumento de la cantidad del aporte calórico positivo por parte de la madre otorgan al feto un aumento drástico de ácidos grasos y glucosa que son transferidos a través del tejido placentario, y debido a que el almacenamiento subcutáneo de lípidos es un evento relativamente tardío en la vida fetal, esto representa una adversidad para el feto al comienzo del embarazo, ya que es incapaz de almacenar grasa subcutánea como amortiguador de almacenamiento, y así manejar el exceso de nutrientes, por lo que almacena grasa en diferentes órganos como el hígado, páncreas y músculo esquelético, lo que lo predispone al desarrollo de resistencia a la insulina y EHGNA14,45,46.
Varios estudios han demostrado que en mujeres embarazadas alimentadas con una ingesta calórica positiva alta en grasas se producen alteraciones en el feto sobre el desarrollo de órganos metabólicos clave como el hígado, donde se instaura estrés oxidante, disfunción mitocondrial, aumento de mediadores proinflamatorios como adipocina y leptina, además de alteración de lipogénesis y apoptosis46-48. Esto altera la expresión genética en las células de Kuppfer y aumenta la expresión del factor de necrosis tumoral alfa (TNFα), lo que promueve posteriormente la progresión a fibrosis y esteatohepatitis, consistente con el desarrollo de EHGNA en la vida temprana y su progresión49-55.
De acuerdo con la DOHaD, la información heredada no codificada en la secuencia del ADN puede regular una variedad de fenotipos histopatológicos característicos de la EHGNA, programados en respuesta a los eventos adversos durante el embarazo como la obesidad y la resistencia a la insulina, los cuales pueden persistir después del nacimiento, a pesar de la ausencia de factores de riesgo como obesidad, resistencia a la insulina o inflamación sistémica o local del tejido adiposo, lo que indica que la programación epigenética para una predisposición a EHGNA puede ser incapaz de revertirse independientemente del régimen alimenticio durante la juventud y adultez56.
Adicionalmente, el aumento de mediadores inflamatorios también modifica la sensibilidad hipotalámica de los reguladores hormonales, clave para el apetito y el metabolismo, que impactan el apetito postnatal relacionado con sobrepeso y obesidad más adelante en la vida57-59.
Un IMC alto al inicio del embarazo aumenta el riesgo para la descendencia de padecer obesidad y trastornos relacionados con esta en la edad adulta60. El entorno obesogénico también aumenta el riesgo de desarrollar resultados adversos neonatales y complicaciones del embarazo, que incluyen alteraciones del crecimiento y desarrollo, hiperglucemia, resistencia a la insulina, niveles altos de HOMA-IR, alteraciones a nivel conductual, cognitivo, reproductivo, defectos a nivel cardiovascular, gastrointestinal, inmunológico, respiratorio, que incluyen desenlaces fatales como parto prematuro y muerte fetal61-66.
Un estudio de casos y controles en ratones comparó la dieta estándar y la dieta obesogénica durante la gestación, teniendo como resultado que la descendencia de madres obesas en comparación con la descendencia de madres delgadas obtuvo una elevación significativa del contenido de triglicéridos del tejido hepático, la puntuación media de esteatosis hepática y el marcador plasmático de aspartato transaminado. Además, paralelamente a la lesión hepática inducida, se reportó un aumento de fibrogénesis demostrada por la expresión diferencial del gen del colágeno 1-α2; se concluyó que la exposición de la descendencia a la obesidad materna durante el embarazo programa un fenotipo característico de EHGNA en las crías, que además afecta críticamente el período posnatal temprano y posiblemente implique una alteración de la señalización de los núcleos del apetito hipotalámicos relacionados con la leptina67.
Perfiles de metilación diferencial asociados a programación fetal en obesidad gestacional
Un metaanálisis de 19 cohortes independientes centrado en la metilación diferencial de ADN en la sangre del cordón umbilical del recién nacido, expuesto a un alto IMC en el embarazo, comparó los pares madre-hijo de mujeres con peso normal (n = 4834) y mujeres con sobrepeso (n = 2885 de las cuales 1299 eran obesas). Un IMC alto materno en el inicio del embarazo se asoció con metilación diferencial en 9,044 sitios CpG que después del ajuste mediante la corrección de Bonferroni para el recuento de células resultó en 86 sitios. Los perfiles de metilación asociados con un IMC materno elevado se asociaron con niveles más bajos de metilación. Los 86 sitios de metilación se relacionaron con 77 regiones genéticas, con varios sitios en el mismo gen: RBMS1 (3 sitios), POM121L1P (3 sitios), VIPR2 (2 sitios), SQLE (2 sitios), RASA3 (2 sitios), MIR200B (2 sitios), KAT6B (2 sitios). A partir de un subgrupo de 4 cohortes, se analizó el seguimiento de los recién nacidos hasta la adolescencia (15 a 18 años). De los 86 sitios reportados, en este subgrupo de 4 cohortes, se encontró 72 sitios que continuaban con metilación diferencial relacionados a los siguientes genes: CDHR3, ACTL10/NECAB3, POM121L1P, VIPR2, AGRN y GGTLC168.
El estudio de la cohorte Newborn the Epigenetics Study (NEST) incluyó 360 pares de madres e hijos. Antes del embarazo, 48% tenían bajo peso (IMC inferior a 25 kg/m2), 21%, sobrepeso (IMC de 25 a 29,99 kg/m2), y 31%, obesidad (IMC ≥30 kg/m2). Los niveles diferenciales de metilación obtenidos de sangre del cordón umbilical resultantes en el grupo con obesidad fueron de 6148 sitios CpG, correspondientes a 1034 genes, de los cuales, después del ajuste de la proporción de células, se redujo el número a 876 sitios, correspondientes a 101 genes. Los genes con diferencias de metilación más robustas que se asociaron con la obesidad materna correspondieron a TAPBP (57 CpG), RP11 (13 CpG), NFE2L3 (6 CpG), GLIPR1L2 (11 CpG) y MFSD1 (10 CpG). La cuantificación de la metilación en el gen TAPBP entre descendientes nacidos de mujeres obesas (n = 20) y no obesas (n = 24) mostró que los descendientes de mujeres obesas tenían aproximadamente un 2% más de metilación que los descendientes de mujeres no obesas69.
Una réplica del estudio NEST con la cohorte del estudio longitudinal de padres e hijos de Avon (ALSPAC) analizó los datos de metilación de 751 parejas de madre e hijo. La obesidad materna previa al embarazo mostró asociación con la metilación diferencial en 10 sitios CpG del gen TAPBP entre la descendencia femenina, y en 12 CpG del mismo gen entre la descendencia masculina. Entre los 10 CpG del gen TAPBP identificados en la descendencia femenina, solo 2 se superpusieron con los identificados en la cohorte NEST, y de los 12 CpG del gen TAPBP identificados en masculinos, ninguno se superpuso en comparación con el estudio NEST. La importancia funcional de los niveles diferenciales de metilación en las determinaciones de los sitios CpG de TAPBP está asociada a la categoría de genes relacionados con riesgo de alteraciones cardiometabólicas69.
Adicionalmente, como medida de análisis comparativo con el consorcio Pregnancy and Childhood Epigenetics (PACE), que incluyó 23 cohortes independientes, donde 77 genes fueron reportados con metilación diferencial, se encontró que principalmente pertenecen a las categorías sobre la expresión de resistencia a la insulina y aumento de los niveles de lipoproteínas en la decendencia69.
De la cohorte ALSPAC, un estudio obtuvo muestras de sangre del cordón umbilical de los hijos de madres con obesidad (n = 32), y se compararon con las madres de peso normal (n= 577). Se reportaron 28 sitios CpG diferencialmente metilados. Las 4 regiones genéticas más diferencialmente metiladas de los hijos de madres obesas en comparación de madres no obesas fueron: SUCLG2, FAM129B, KIF15 y STAB2. De estos, se reportó mayor metilación con los sujetos entre 0 a 7 años en comparación con menor metilación cuando alcanzaron el rango entre los 7 y 17 años70.
Un estudio de casos y controles enfocado a la caracterización de los sistemas de adipocinas (ligandos y receptores) reclutó 30 mujeres entre 27 y 37 años con embarazo único y parto por cesárea entre las 37 a 41 semanas de gestación, asignadas con un IMC en el primer trimestre de 18-25 kg/m2 en el grupo control y con 30-40 kg/m2 en el grupo obeso. Las muestras se recolectaron de placenta del lado fetal. Los niveles de metilación diferencial del promotor del gen leptina (LEP) reportaron 17 sitios CpG, de los cuales 7 CpG estaban hipometilados y 4 estaban hipermetilados; el nivel medio de metilación del ADN fue significativamente mayor en el grupo de obesos que en el grupo control. Por lo que el sistema de la leptina en la obesidad materna parece estar asociado con una elevada metilación del ADN del promotor del gen LEP en el lado fetal de la placenta. Los niveles de metilación diferencial de la región promotora de ADIPOQ de 2 regiones (21 sitios CpG para la región 1 y 12 de la región 2) reportaron que estaban hipometiladas (<5%), mientras que para los niveles de metilación del ADN en la región promotora ADIPOR1 (21 sitios CpG) y la región promotora ADIPOR2 (16 sitios CpG) también estaban hipometiladas (<5%). En conclusión, este estudio mostró una metilación diferencial de leptina y adiponectina como una respuesta adaptativa placentaria a la obesidad materna (ver Figura 2)71.
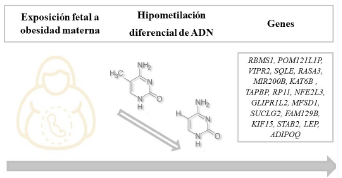
Fuente: tomado y editado de Nogues P, et al.68,69,70,71
Figura 2 Perfiles de metilación diferencial de ADN en exposición fetal a la obesidad gestacional. La obesidad materna promueve la expresión de perfiles con mayor o menor nivel de metilación diferencial del ADN en secuencias CpG promotoras de distintos genes en sus secuencias subyacentes.
Conclusiones
Los procesos exactos de cómo funciona la programación fetal aún no han sido comprendidos en su totalidad, aunque sí se han confirmado efectos en la descendencia mediante la regulación de los mecanismos epigenéticos. Los procesos de metilación diferencial de distintos estudios recabados reportan que en las mujeres embarazadas alimentadas con una ingesta calórica positiva alta en grasas se producen alteraciones en el feto sobre el desarrollo de órganos metabólicos clave como el hígado, lo que puede predisponer al desarrollo de EHGNA pediátrica. Los análisis posteriores a los resultados obtenidos de los distintos perfiles de metilación diferencial mencionados señalan que las categorías metabólicas a las que pertenecen se asocian con alteraciones cardiometabólicas, resistencia a la insulina, lipoproteínas y disfunción de las hormonas adiponectina y leptina.
A pesar de los avances expuestos, aún queda un largo camino por recorrer, ya que la posibilidad de obtener nuevos biomarcadores por parte de la epigenética para el diagnóstico y seguimiento de la EHGNA podría brindar la oportunidad de prevenir la EHGNA pediátrica, así como también su posible progresión a lo largo de la vida.

