










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Universidad Industrial de Santander. Salud
Print version ISSN 0121-0807On-line version ISSN 2145-8464
Rev. Univ. Ind. Santander. Salud vol.42 no.1 Bucaramanga Jan./Apr. 2010
polimorfismos del gen de la enzima
óxido nítrico sintasa endotelial
Norma Cecilia Serrano1, Luis Alfonso Díaz1, María Carolina Páez1, Juan Pablo Casas2, Juan Pablo Casas3
1. Centro de Investigaciones Biomédicas, Universidad Autónoma de Bucaramanga, Bucaramanga, Colombia.
2. Department of Epidemiology and Public Health, University College London, London, WC1E 6BT, United Kingdom.
3. Department of Epidemilogy and Population Health, London School of Hygiene and Tropical Mediane (LSHTM), London,
WC1E 7HT, United Kingdom.
Correspondencia: Norma Cecilia Serrano Díaz, MD MSc. Centro de Investigaciones Biomédicas, Universidad Autónoma de
Bucaramanga, Bucaramanga, Colombia. Campus el Bosque, Calle 157 N° 19-55 Cañaveral Parque, Bucaramanga - Colombia.
E-mail: nserrano@unab.edu.co. Fax: 577-6399147. Teléfono: 577-6399152, Ext. 139.
Recibido: 12 de octubre de 2009 - Aceptado: 14 de diciembre de 2009
RESUMEN
El óxido nítrico (NO) juega un papel importante en la regulación de la homeostasis vascular. La producción endotelial de NO está regulada por la óxido nítrico sintasa endotelial (NOSe), por lo tanto variaciones genéticas en el gen NOS3 podrían influir en la producción de NO. Tres polimorfismos (Glu298Asp, intrón-4 y -786T>C) en el gen NOS3, han sido asociados de forma inconsistente con enfermedades cardiovasculares (ECV). Estas variantes genéticas se han asociado con disminución de RNAm, concentración séricas bajas de nitritos/nitratos y disminución de la reactividad endotelial. A pesar de la amplia investigación de estos polimorfismos en ECV y de las aproximaciones para evaluar el papel funcional, no existe evidencia suficiente que permita esclarecer el papel causal y funcional de dichos polimorfismos sobre la enfermedad. Se requiere de nuevas estrategias que permitan seleccionar polimorfismos funcionales para determinar el riesgo atribuido al genotipo sobre la enfermedad. En la presente revisión se discute el posible efecto sobre la expresión de la actividad de la NOSe de esto tres polimorfismos genéticos descritos como de alta relevancia clínica en enfermedades cardiovasculares. Salud UIS 2010; 42: 66-77
Palabras Claves: Óxido nítrico, óxido nítrico sintasa, Polimorfismos Genéticos, polimorfismo de nucleótido simple, enfermedad cardiovascular
Functional relevance of polymorphisms of
endothelial nitric oxide synthase gene
ABSTRACT
Nitric oxide (NO) plays an important role on vascular homeostasis regulation. NO endothelial production is regulated by endothelial nitric-oxide synthase (eNOS), reason for which genetic variations in NOS3 gene could influence NO production. Three polymorphisms (Glu298Asp, intron-4, and -786T>C) in NOS3 gene have been inconsistently associated with cardiovascular diseases (CVD). These genetic variables have been linked to diminishment mRNA, nitrites/nitrate low serum levels, and low endothelial reactivity. Despite wide research on these polymorphisms in CVD and several approaches to evaluate their functional role, not enough evidence is available to clarify their functional and causal participation over the disease. New strategies are required to select functional polymorphisms and to determine genotype-attributable risk over disease. In this paper, we review and discuss the possible effect on NOSe activity expression of these genetic polymorphisms with high clinical relevance in CVD diseases. Salud UIS 2010; 42: 66-77.
Keywords: Nitric oxide, nitric oxide synthase, Genetic Polymorphisms, single nucleotide polymorphism, cardiovascular disease
INTRODUCCIÓN
El óxido nítrico (NO) juega un papel importante en la regulación de la homeostasis vascular1, siendo un potente vasodilatador endógeno; además, inhibe la agregación plaquetaria2, atenúa la adhesión de leucocitos al endotelio3, modula la proliferación del músculo liso4, 5 e inhibe la oxidación del colesterol de baja densidad (LDL)6, 7. El NO es producido en el endotelio vascular por medio de la óxido nítrico sintasa endotelial (NOSe), por acción calcio-calmodulina dependiente, a partir de la conversión de L-arginina a L-citrulina y NO8.
El óxido nítrico es un gas simple que no se almacena, y se libera una vez se sintetiza. La regulación en la generación de NO puede ocurrir por alteraciones en la expresión o actividad de la NOSe, por cambios en la disponibilidad de cofactores activadores o por la producción de moléculas endógenas inhibitorias9-12. El papel protagónico que juega el NO sobre el endotelio vascular ha llevado a proponer que una deficiencia vascular de NO, ya sea hereditaria o adquirida, podría contribuir fuertemente a disfunción endotelial y, en consecuencia, al desarrollo de enfermedades cardiovasculares.
La enzima NOSe es codificada por el gen NOS3, localizado en el cromosoma 7q35-36, el cual fue clonado y secuenciado en 1993. Está conformado por 26 exones que codifican para una proteína de 135 kD con 1203 aminoácidos. Una secuencia de aproximadamente 1500 bp corriente arriba de la región promotora del gen contiene sitios de unión a factores de transcripción que median la regulación por estrés de fricción y estrógenos entre otros13.
Dado que la regulación en la disponibilidad del NO en el endotelio está determinada en gran medida por la síntesis de la NOSe; variaciones genéticas (polimorfismos) en NOS3 son potenciales candidatos para ser estudiados como determinantes de la biodisponiblidad de NO y por ende, relacionados con disfunción endotelial. Por lo tanto, polimorfismos en NOS3 podrían explicar deficiencias funcionales, cuantitativas (alteraciones de la transcripción, estabilidad del RNAm, formación de una proteína con defectos catalíticos) o incremento en la degradación de la enzima14. Dichos polimorfismos podrían encontrarse con una frecuencia significativamente mayor en individuos con patologías que cursan con disfunción endotelial tales como, aterosclerosis14, enfermedad arterial coronaria15, 16, hipertensión arterial17 o preeclampsia18, entre otras.
En la presente revisión se discute, a la luz del conocimiento actual, el posible efecto sobre la expresión, actividad de enzima y niveles de NO, de tres polimorfismos genéticos de la NOSe, que han sido descritos como de alta relevancia clínica en enfermedades cardiovasculares.
Polimorfismos Genéticos
Desde la terminación del secuenciamiento del genoma humano19, 20, se han dado grandes pasos en la descripción y clasificación de las variaciones en los genes. La variación genética más común en los humanos es el polimorfismo de nucleótido simple (SNP, por su nombre en inglés, single nucleotide polymorphism). La aparición de un SNP dentro del genoma humano es bastante común, aproximadamente uno cada 180 pb21. Actualmente hay más de 10 millones de SNP documentados y almacenados para uso público en bases de datos libres, tales como www.hapmap.org. Aunque la aparición de SNPs dentro del genoma humano es bastante frecuente, la mayoría de ellos tienen una distribuciòn de alelos poco informativa (64% de todos los SNP), con una frecuencia del alelo menor (MAF) de menos del 5%. Además pocos SNP se ubican en las regiones codificantes del gen [(cSNP) 4%], comparado con los ubicados en las regiones no codificantes que corresponden al 96%. Para un gen promedio, se calcula que pueden existir cerca de 126 SNP, de los cuales el 35% corresponden a SNP comunes (MAF≥5%) y tan solo el 4% serían cSNP21. Estas observaciones son importantes al momento de escoger los SNP de los genes candidatos involucrados en los estudios de asociación. Con la disponibilidad actual de un catálogo de SNP comunes, los investigadores han desarrollado métodos para aplicar los SNP a los estudios de asociación con el objetivo de identificar variaciones del DNA que contribuyan a incrementar la susceptibilidad para enfermedades humanas.
Para NOS3 se han descrito más de 160 SNP, 8% de ellos ubicados en regiones codificantes, un poco mayor a lo reportado para el genoma en general, y un 92% localizado en regiones no codificantes (Figura 1). Cada uno de ellos con frecuencias alélicas que difieren entre los grupos étnicos (GE) y poblaciones (P) en las cuales son captados los individuos para la genotipificación y definición de las frecuencias alélicas. Por ejemplo, para el GE: Caucásicos, P: residentes de Utah con ascendencia del norte y occidente de Europa; GE: Asiáticos, P: Chinos y Japoneses; GE: Africanos, P: Sub-Sahariano, Nigeria, etc. Para fines de este articulo, utilizamos como referencia frecuencias que han sido publicadas en el GE de caucásicos.
Utilizando bases de datos electrónicas disponibles para la identificación de SNP, realizamos una búsqueda sistemática para NOS3 con base en la localización e implicación molecular del SNP, estado de validación, MAF y heterocigocidad.
Localización e implicaciones moleculares
Se identifica los SNP al interior de las diferentes regiones del gen y con base en su rol funcional se seleccionan los de interés, principalmente los no sinónimos ubicados en regiones codificantes y promotora, y algunos en la unidad terminal de la región UTR, (Tabla 1).22.
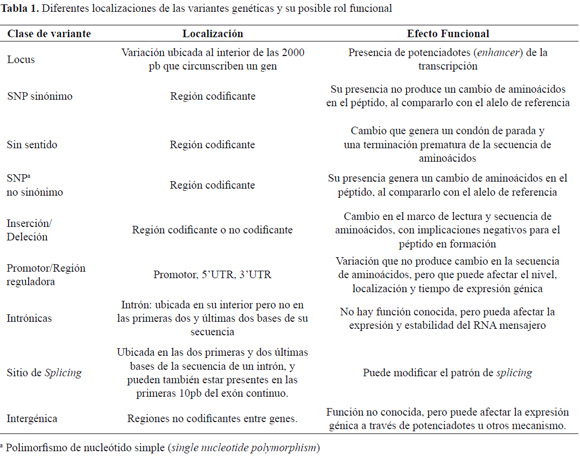
Estado de validación
Hace alusión a los diferentes análisis realizados para validar una variante genética recientemente reportada, que incluye el ser sometido de manera independiente por más de un grupo, ser validado por frecuencia con la respectiva identificación del alelo menor en diferentes poblaciones y ser evaluado por el Proyecto Internacional HapMap23. Para el caso particular de NOS3 el porcentaje de SNP validados por este consorcio no supera el 7% (Tabla 2).

Frecuencia del alelo menor (MAF)
Valor que describe la frecuencia del alelo no común en la población de referencia, que en el caso de estudios de asociación seria el "alelo de riesgo", y cuyo porcentaje nos permite predecir el tamaño de muestra necesario para evaluar el alelo en determinado grupo poblacional. En la (Tabla 3), se identifica el total de SNP de NOS3 con un valor MAF ≥10% en población caucásica.
Heterocigocidad
Reporte del genotipo en su versión heterocigoto para el SNP de interés, en más del 30% de la población de referencia. A pesar de ser un gen con un número alto de polimorfismos, solo tres de ellos se han evaluado rutinariamente en estudios de asociación genética y se han considerado de relevancia clínica, especialmente en enfermedades cardiovasculares. Un SNP ubicado en la región promotora del gen, a -786 pb del sitio de inicio de la transcripción (-786C>T), un VNTR al interior del intrón-4 y un SNP no sinónimo ubicado en el exón 7 (Figura 2). Sin embargo la variación funcional que genera cada uno estos polimorfismos no está aún completamente caracterizada, por lo tanto los estudios de asociación están basados más en "polimorfismos candidatos", que en "genes candidatos" 24.

Función de los Polimorfismos
Glu298Asp. Este polimorfismo se ubica en el exón 7 del gen, cuya variante 894 G>T, da como resultado un cambio del aminoácido glutamato por ácido aspártico en la posición 298 (Glu298Asp) de la proteína. Estudios de asociación genética han mostrado que individuos portadores del alelo Asp298 tienen una probabilidad mayor de desarrollar enfermedad arterial coronaria15, 16, preeclampsia18, espasmo de la arteria coronaria25 e hipertensión arterial17, 26; sin embargo, los resultados de asociación han sido inconsistentes cuando se replican en diferentes poblaciones.
Dado que los cambios de glutamato por ácido aspártico son sustituciones conservativas, inicialmente se consideró que el polimorfismo Glu298Asp estaba en desequilibrio de ligamiento con otro polimorfismo funcional no identificado. Sin embargo, resultados in vitro recientes indican que el polimorfismo Glu298Asp por sí mismo da como resultado una alteración en la función de la enzima. Es así, como Tesauro et al27 demostraron in vitro que las isoformas de la NOSe son procesadas de manera diferente dependiendo de la presencia de ácido aspártico o glutamato en la posición 298. Cuando la NOSe tiene en la posición 298 ácido aspártico, ésta es clivada en la vecindad del aminoácido 298, lo cual no sucede en presencia del aminoácido glutamato, indicando un cambio potencial en la estructura secundaria de la proteína determinado por un reemplazo conservativo; por lo tanto, el polimorfismo Glu298Asp podría ser no silente y, posiblemente, ser reconocido por proteasas endógenas como un blanco de clivaje, lo que genera disminución en la biodisponibilidad de NOSe y como consecuencia de NO. Sin embargo, un estudio posterior mostró que posiblemente este clivaje (ex vivo) en presencia de Asp298, podría ser debido a condiciones de incremento de temperatura y pH bajo, dado que éste fue eliminado al usar el mismo sistema de buffer pero limitando la hidrólisis ácida de los enlaces Asp; estos resultados estarían en contra de un procesamiento intracelular diferencial en presencia de Asp o Glu28.
Un estudio posterior, Dosenko29 trató de determinar las manifestaciones funcionales de este polimorfismo, encontró que los niveles de RNAm y la actividad de la enzima en plaquetas dependen del genotipo. Plaquetas procedentes de individuos homocigotos para Asp298 presentan niveles más bajos de RNAm que los homocigotos para el alelo silvestre Glu298, pero los valores no muestran diferencia entre los individuos heterocigotos. La actividad de la enzima medida por un sistema de detección fluorimétrico (FCANOS 1, Sigma), mostró que los homocigotos para Asp298 tenían una menor actividad de NOSe que los individuos homocigotos para el alelo silvestre, pero esta diferencia no fue estadísticamente significativa29. Otros ensayos realizados en plaquetas procedentes de pacientes homocigotos pora el alelo Asp298 mostraron que éstos producen una cantidad mayor de superóxido y menor de NO ex vivo que individuos homocigotos para la variante silvestre Glu29830. A pesar de estos resultados, el efecto in vivo que produce este polimorfismo sobre la producción de NO, estrés oxidativo y la respuesta vascular aún no es claro.
Estudios en muestras pequeñas en las que se utilizó vasodilatación mediada por flujo (VMF), una prueba no invasiva que permite medir la respuesta endotelial dependiente de NO in vivo, utilizando ultrasonido del alta resolución31, han mostrado resultados contradictorios32-36. Un ensayo adicional pero con un tamaño de muestra mayor, conducido por Leeson et al 37, en 248 individuos (131 mujeres y 117 hombres) con edad entre 20 y 28 años, encontró que el polimorfismo Glu298Asp no influye sobre la reactividad endotelial medida por VMF. Sin embargo, varones portadores del alelo Asp298 presentaban una reducción en la reactividad endotelial asociada al estado de tabaquismo, comparado con los individuos fumadores y portadores del alelo Glu298. Estos hallazgos sugieren una modulación determinada genéticamente sobre fenotipo analizado y una clara interacción entre genotipo (Asp298) y factores de riesgo convencionales (tabaquismo)37. Estos hallazgos abren la posibilidad de entrar a realizar intervenciones dirigidas (no fumar), sobre poblaciones específicas a riesgo seleccionadas por marcadores genéticos (portadores del alelo Asp298). Posteriormente, un estudio con un tamaño de muestra significativamente mayor, el cual incluyó 2883 participantes del estudio Framinghan, confirmó la no relación entre el genotipo Glu298Asp y la VMF 38.
Polimorfismo –786T>C. Este polimorfismo se ubica en la región promotora del gen. Fue descrito inicialmente en 1999 por Nakayama et al, quienes encontraron que el alelo mutado, -786C, se asociaba con una marcada reducción en la actividad promotora del gen, 52 ± 11%. Hallazgos que sugerían compromiso en la síntesis endotelial de NO y predisponían a espasmo coronario en población japonesa39. Estudios posteriores confirmaron que en homocigotos para el alelo mutado, C/C, se reducía significativamente la actividad promotora del gen y, por ende, los niveles de RNAm de la NOSe40-42. Sin embargo, otros estudios realizados en población australiana41 y francesa44 encontraron que este polimorfismo no se asocia con la actividad promotora del gen ni con enfermedad arterial coronaria.
Los resultados positivos entre este genotipo y reducción en el RNAm de la NOSe, condujeron a plantear la posible asociación de este polimorfismo con bajos niveles de NO endotelial. Sin embargo, la cuantificación directa de los niveles de NO es difícil, dado su corta vida media; por lo tanto, se ha utilizado como medida indirecta la cuantificación en plasma de nitritos y nitratos (NOx), productos de la rápida oxidación del NO. La medición de NOx en plasma a partir de sangre recolectada posterior a 12 horas de ayuno reduce en cerca de un 50% los niveles de NOx y refleja en gran medida la producción endógena de NO45, 46.
Los resultados con respecto a la correlación entre fenotipo (niveles de NOx) y genotipo (polimorfismo –786T>C) han sido contradictorios. Se ha reportado bajos niveles en las concentraciones del NOx plasmáticas en individuos portadores del alelo "C"47, mientras que otros estudios no muestran diferencias significativas48-50. Aunque este polimorfismo puede reducir la expresión del gen que codifica para la NOSe, aparentemente no afecta las concentraciones plasmáticas de NOx; por lo tanto, parece no tener efecto sobre la producción endógena de NO.
El efecto in vivo de este polimorfismo también ha mostrado resultados contradictorios. Un estudio encontró una relación entre el polimorfismo –786T>C y la VMF durante la infusión de acetilcolina en pacientes hipertensos33; sin embargo otros dos estudios no encontraron dicha correlación34, 35, 38.
VNTR intrón-4. Este polimorfismo corresponde a la una repetición de 27 pb localizado en el intrón 4. El alelo silvestre (4b) está conformado por la repetición de 5 veces consecutivas de la secuencia de 27 pb, mientras que el alelo mutado (4a) presenta 4 veces la secuencia repetida. Aunque este polimorfismo se ubica en un intrón, esto no necesariamente excluye la posibilidad de tener un papel funcional de relevancia en la regulación trans y postranscripcional del gen.
Algunos reportes indican que esta variante podría tener efecto funcional. Wang et al51 presentan en un estudio con individuos japoneses sanos y no fumadores, que las personas homocigotas para el alelo mutado (4a/4a) presentaban un incremento en los niveles plasmáticos de NOx. Esto contrasta con otro estudio, realizado también en población japonesa, donde encuentra niveles disminuidos de NOx en individuos portadores del alelo mutado, pero en este estudio no se documenta el antecedente de tabaquismo52.
Dado que el genotipo 4a/4a ha sido asociado con incremento del riesgo para enfermedad arterial coronaria53, este genotipo debería estar relacionado más con niveles bajos de NOx que con niveles elevados. Una explicación a esta aparente incongruencia está dada por la posible interacción significativa entre tabaquismo y este polimorfismo en enfermedad arterial coronaria. En fumadores, pero no en individuos no fumadores, los autores encuentran un exceso de homocigotos para el alelo mutado (4a/4a) en pacientes con arterias severamente estenosadas, comparado con aquellos sin o con estenosis moderada. Sin embargo, paradójicamente estos individuos (4a/4a) también tenían significativamente niveles más altos de NO que individuos sanos, no fumadores, portadores de uno o dos alelos silvestres (4a/4b, 4b/4b) 53.
Un estudio posterior54, realizado en placentas, encontró que los niveles de RNAm y de la proteína fueron significativamente más bajos en heterocigotas (4a/4b), que en homocigotas para el alelo silvestre. En contraste, la actividad de la enzima fue cerca de 5 veces más alta en las heterocigotas que en las homocigotas (4b/4b), lo cual es consistente con el hallazgo anterior donde asocian el alelo mutado con alta producción de NO53. Sin embargo, en placentas procedentes de fumadoras, se encontró disminución en los niveles de la proteína NOSe, tanto para portadoras del alelo silvestre como del mutado, pero solo para el genotipo 4a/4b, la actividad de la enzima estaba reducida a la mitad de las no fumadoras. Por lo tanto, aunque portadores del alelo mutado producen más cantidad de NO, la capacidad generadora de NO por la NOSe está seriamente comprometida en fumadores portadores del alelo mutado más no en aquellos portadores del alelo silvestre54. Entonces, parece factible hipotetizar que el tabaquismo regula la actividad de la enzima NOSe dependiendo de la variante ubicada en el intrón 4, ejerciendo un efecto negativo en presencia del alelo mutado.
El efecto in vivo de este polimorfismo ha sido documentado en un estudio conducido en pacientes con diabetes 2, en el cual se encontró que los pacientes diabéticos y fumadores portadores del alelo 4a presentaban una reducción en la VMF en fumadores pero no en los pacientes no fumadores55.
Por otro lado, un estudio realizado in vitro, para evaluar la relación entre genotipo-fenotipo, en el que se utilizó cultivo de células endoteliales procedente de vena de cordón umbilical expuestas a extracto de cigarrillo, mostró que para ninguno de los tres polimorfismos de la NOSe (Glu298Asp, –786T>C, VNTR intron-4) existe una correlación entre el genotipo y la exposición a extracto de cigarrillo. Estos hallazgos podrían indicar que la aparente asociación entre tabaquismo y genotipo NOSe, podría darse a nivel de interacción biológica funcional más que en la expresión de NOSe o en la actividad de la enzima. Una posibilidad es que los radicales libres producidos por el fumar cigarrillo pudiesen interferir con la producción biológica de NO por la NOSe56.
CONCLUSIONES Y
RECOMENDACIONES PARA
FUTURAS INVESTIGACIONES
Sin duda la NOSe es una enzima importante para la integridad fisiológica de la pared arterial y se encuentra involucrada en el desarrollo de disfunción endotelial. También parece claro que las variaciones genéticas de NOSe pueden llevar a cambios en la expresión y actividad de la enzima, los cuales podrían ser modificados por factores ambientales; sin embargo, los estudios funcionales tanto in vitro como in vivo han generado datos contradictorios. Las asociaciones aparentemente inconsistentes entre genotipos de la NOSe y fenotipos clínicos (hipertensión, espasmo coronario, preeclampsia o enfermedad arterial coronaria entre otros), o con fenotipos bioquímicos (expresión y actividad de NOSe, niveles séricos de NOx y VMF), es un fenómeno observado en muchos otros genes y fenotipos clínicos, fenómeno que no solo ocurre en enfermedades complejas sino también en enfermedades Mendelianas aparentemente "simples" (monogénicas).
La asociación entre polimorfismos genéticos y fenotipos clínicos varían cuantitativa y cualitativamente para la misma mutación, así como también entre la misma familia, la misma población o poblaciones. Aunque el mecanismo para explicar dicha variación entre la asociación genotipo-fenotipo no es clara aún, Dipple and McCabe57, han presentado un modelo de umbral para tratar de explicar dicha inconsistencia. Factores tales como los ambientales, alelos independientes, interacción gen-gen y variabilidad en la severidad del fenotipo clínico, pueden todos ellos potencialmente modificar o cambiar la relación genotipo-fenotipo a otro tipo de relación, particularmente la severidad de la enfermedad.
Hasta el momento, se ha acumulado una cantidad considerable de evidencia que evalúa el papel del gen del la NOSe y sus variables genéticas tanto en enfermedades cardiovasculares como en otros desórdenes complejos. No obstante, el papel funcional de los diferentes polimorfismos de la NOSe, no es aún claro, en parte debido a la complejidad técnica en la cuantificación de la producción de NO en humanos, pero más aún, en la ausencia de estrategias claras que permitan definir "polimorfismos funcionales".
A la fecha se acepta que la evaluación de genes candidatos como el de la NOSe, requiere una estrategia integral que involucre la evaluación de todas las variantes presentes en el gen, teniendo en cuenta el grado de vínculo [desequilibrio de ligamiento (DL)] presente entre ellas. Es así, como gracias a la rápida evolución del Proyecto Genoma Humano19,20 y otras estrategias como el Proyecto HapMap58, es ahora posible realizar este tipo de evaluaciones, seleccionando dentro de las regiones del alto DL los SNP blanco a evaluar (tSNP), en cuya selección se recomienda incluir solo variantes con una frecuencia del alelo menor >5%59-62. Finalmente los tSNP seleccionados y los haplotipos generados por los tSNP, serán genotipificados en la población a estudio, en un tamaño de muestra adecuado que permita determinar la fuerza de asociación con el fenotipo clínico (enfermedad) o bioquímico (expresión, actividad, niveles de la proteína)38.
A pesar de la amplia investigación en torno al gen la NOSe en enfermedades cardiovasculares y otros desórdenes complejos, y de las aproximaciones para evaluar el papel funcional de los polimorfismos estudiados, no existe evidencia suficiente que permita esclarecer el papel causal de dichos polimorfismos sobre la enfermedad, ni entender la naturaleza de la relación genotipo-fenotipo y la interacción entre genotipo y medio ambiente. Por lo tanto se requiere aún de nuevos estudios, utilizando nuevas estrategias que permitan determinar el riesgo atribuido al genotipo para construir estrategias de intervención dependientes del genotipo y dependientes de la interacción genotipo-ambiente.
AGRADECIMIENTOS
La presente revisión fue realizada con patrocinio de Colciencias (proyecto 1241-04-13030) y de la Universidad Autónoma de Bucaramanga (proyecto EGEN07).
CONFLICTO DE INTERÉS
Los autores declaran no tener ningún conflicto de intereses en la realización del presente trabajo.
REFERENCIAS
1. Radomski MW, Palmer RM, Moncada S. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet 1987; 2: 1057-1058. [ Links ]
2. Bath PM, Hassall DG, Gladwin AM, Palmer RM, Martin JF. Nitric oxide and prostacyclin. Divergence of inhibitory effects on monocyte chemotaxis and adhesion to endothelium in vitro. Arterioscler Thromb 1991; 11: 254-260. [ Links ]
3. Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA 1991; 88: 4651-4655. [ Links ]
4. Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest 1989; 83:1774-1777. [ Links ]
5. Sarkar R, Meinberg EG, Stanley JC, Gordon D, Webb RC. Nitric oxide reversibly inhibits the migration of cultured vascular smooth muscle cells. Circ Res 1996; 78:225-230. [ Links ]
6.Hogg N, Kalyanaraman B, Joseph J, Struck A, Parthasarathy S. Inhibition of low-density lipoprotein oxidation by nitric oxide. Potential role in atherogenesis. FEBS Lett 1993; 334: 170-174. [ Links ]
7. Nakane M, Mitchell J, Forstermann U, Murad F. Phosphorylation by calcium calmodulin-dependent protein kinase II and protein kinase C modulates the activity of nitric oxide synthase. Biochem Biophys Res Commun 1991; 180: 1396-1402. [ Links ]
8. Knowles RG, Moncada S. Nitric oxide synthases in mammals. Biochem J 1994; 298: 249-258. [ Links ]
9. Charles IG, Scorer CA, Moro MA, Fernández C, Chubb A, Dawson J, et al. Expression of human nitric oxide synthase isozymes. Methods Enzymol 1996; 268: 449-460. [ Links ]
10. Vallance P. Exploring vascular nitric oxide in health and disease. The Goulstonian Lecture 1996. J R Coll Physicians Lond 1997; 31: 321-327. [ Links ]
11. Calver A, Collier J, Leone A, Moncada S, Vallance P. Effect of local intra-arterial asymmetric dimethylarginine (ADMA) on the forearm arteriolar bed of healthy volunteers. J Hum Hypertens 1993; 7:193-194. [ Links ]
12. Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992; 339: 572-575. [ Links ]
13. Marsden PA, Heng HH, Scherer SW, Stewart RJ, Hall AV, Shi XM, et al. Structure and chromosomal localization of the human constitutive endothelial nitric oxide synthase gene. J Biol Chem 1993; 268: 17478-17488. [ Links ]
14. Hingorani AD. Polymorphisms in endothelial nitric oxide synthase and atherogenesis: John French Lecture 2000. Atherosclerosis 2001; 154: 521-527. [ Links ]
15. Hingorani AD, Liang CF, Fatibene J, Lyon A, Monteith MS, Parsons A, et al. A common variant of the endothelial nitric oxide synthase Glu298Asp is a major risk factor for coronary artery disease in the UK. Circulation 1999; 100:1515-1520. [ Links ]
16. Casas JP, Bautista LE, Humphries SE, Hingorani AD. Endothelial nitric oxide synthase genotype and ischemic heart disease meta-Analysis of 26 studies involving 23028 subjects. Circulation 2004; 109:1359-1365. [ Links ]
17. Sandrim VC, Coelho EB, Nobre F, Arado GM, Lanchote VL, Tanus-Santos JE. Susceptible and protective eNOS haplotypes in hypertensive black and white subjects. Atherosclerosis 2006; 186: 428-432. [ Links ]
18. Serrano NC, Casas JP, Díaz LA, Páez C, Mesa CM, Cifuentes R, et al. Endothelial NO synthase genotype and risk of preeclampsia: a multicenter case-control study. Hypertension 2004; 44: 702-707. [ Links ]
19. International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001; 409:860-921. [ Links ]
20. Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science 2001; 291:1304-1351. [ Links ]
21. Crawford DC, Akey DT, Nickerson DA. The patterns of natural variation in human genes. Annu Rev Genomics Hum Genet 2005; 6: 287-312. [ Links ]
22. Tabor HK, Rish NJ, Myers RM. Candidate-gene approaches for studying complex genetic traits: practical considerations. Nature Rev 2002; 3:1-7. [ Links ]
23. The International HapMap Project. Nature 2003; 426:789-796. [ Links ]
24. Goldstein DB. Pharmacogenetics in the laboratory and the clinic. N Engl J Med 2003; 348: 553-556. [ Links ]
25. Yoshimura M, Yasue H, Nakayama M, Shimasaki Y, Sumida H, Sugiyama S, et al. A missense Glu298Asp variant in the endothelial nitric oxide synthase gene is associated with coronary spasm in the Japanese. Hum Genet 1998; 103: 65-69. [ Links ]
26. Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995; 377: 239-242. [ Links ]
27. Tesauro M, Thompson WC, Rogliani P, Qi L, Chaudhary PP, Moss J. Intracellular processing of endothelial nitric oxide synthase isoforms associated with differences in severity of cardiopulmonary diseases: cleavage of proteins with aspartate vs. glutamate at position 298. Proc Natl Acad Sci USA 2000; 6: 2832-2835. [ Links ]
28. Fairchild TA, Fulton D, Fontana JT, Gratton JP, McCabe TJ, Sessa WC. Acidic hydrolysis as a mechanism for the cleavage of the Glu(298)-->Asp variant of human endothelial nitric-oxide synthase. J Biol Chem 2001; 276: 26674-26679. [ Links ]
29.Dosenko VE, Zagoriy VY, Haytovich NV, Gordok OA, Moibenko AA. Allelic polymorphism of endothelial NO-synthase gene and its functional manifestations. Acta Biochim Pol 2006; 53: 299-302. [ Links ]
30. Desantis P, Babaoglu MO, Pezzullo JC, Abermethy DR, Freedman JE. Impaired production of platelet-derived nitric oxide in human subjects with a polymorphic variant of endothelial nitric oxide synthase [abstract]. Circulation 2000; 102(Suppl 2): S61. [ Links ]
31. Celermajer DS, Sorensen KE, Gooch VM, Spiegelhalter DJ, Miller OI, Sullivan ID, et al. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet 1992; 340:1111-1115. [ Links ]
32. Komatsu M, Kawagishi T, Emoto M, Shoji T, Yamada A, Sato K, et al. ecNOS gene polymorphism is associated with endothelium-dependent vasodilatation in type 2 diabetes. Am J Physiol 2002; 283: 557- 561. [ Links ]
33. Rossi GP, Taddei S, Virdis A, Cavallin M, Ghiadoni L, Favilla S, et al. The T-786C and Glu298Asp polymorphisms of the endothelial nitric oxide gene affect the forearm blood flow responses of Caucasian hypertensive patients. J Am Coll Cardiol 2003; 41:938-945. [ Links ]
34. Li R, Lyn D, Lapu-Bula R, Oduwole A, Igho-Pemu P, Lankford B, et al. Relation of endothelial nitric oxide synthase gene to plasma nitric oxide level, endothelial function, and blood pressure in African Americans. Am J Hypertens 2004; 17: 560-567. [ Links ]
35. Paradossi U, Ciofini E, Clerico A, Botto N, Biagini A, Colombo MG. Endothelial function and carotid intima-media thickness in young healthy subjects among endothelial nitric oxide synthase Glu2983Asp and T-7863C polymorphisms. Stroke 2004; 35: 1305-1309. [ Links ]
36. Naber CK, Baumgart D, Altmann C, Siffert W, Erbel R, Heusch G. eNOS 894T allele and coronary blood flow at rest and during adenosine-induced hyperemia. Am J Physiol Heart Circ Physiol 2001; 281: H1908-1912. [ Links ]
37. Leeson CP, Hingorani AD, Mullen MJ, Jeerooburkhan N, Kattenhorn M, Cole TJ, et al. Glu298Asp endothelial nitric oxide synthase gene polymorphism interacts with environmental and dietary factors to influence endothelial function. Circ Res 2002; 90: 1153-1158. [ Links ]
38. Kathiresan S, Larson MG, Vasan RS, Guo CY, Vita JA, Mitchell GF, et al. Common genetic variation at the endothelial nitric oxide synthase locus and relations to brachial artery vasodilator function in the community. Circulation 2005; 112:1419-1427. [ Links ]
39. Nakayama M, Yasue H, Yoshimura M, Shimasaki Y, Kugiyama K, Ogawa H, et al. T-786-->C mutation in the 5'-flanking region of the endothelial nitric oxide synthase gene is associated with coronary spasm. Circulation 1999; 99: 2864-2870. [ Links ]
40. Miyamoto Y, Saito Y, Nakayama M, Shimasaki Y, Yoshimura T, Yoshimura M, et al. Replication protein A1 reduces transcription of the endothelial nitric oxide synthase gene containing a -786T-->C mutation associated with coronary spastic angina. Hum Mol Genet 2000; 9: 2629-2637. [ Links ]
41. Wang XL, Wang J. Endothelial nitric oxide synthase gene sequence variations and vascular disease. Mol Genet Metab 2000; 4: 241-251. [ Links ]
42. Bilsborough W, Green DJ, Mamotte CD, van Bockxmeer FM, O'Driscoll GJ, Taylor RR. Endothelial nitric oxide synthase gene polymorphism, homocysteine, cholesterol and vascular endothelial function. Atherosclerosis 2003; 169: 131-138. [ Links ]
43. Sim AS, Wang J, Wilcken D, Wang XL. MspI polymorphism in the promoter of the human endothelial constitutive NO synthase gene in Australian Caucasian population. Mol Genet Metab 1998; 65: 62-64. [ Links ]
44. Poirier O, Mao C, Mallet C, Nicaud V, Herrmann SM, Evans A, et al. Polymorphisms of the endothelial nitric oxide synthase gene - no consistent association with myocardial infarction in the ECTIM study. Eur J Clin Invest 1999; 29: 284-290. [ Links ]
45. Rosselli M, Imthurn B, Keller PJ, Jackson EK, Dubey RK. Circulating nitric oxide (nitrite/nitrate) levels in postmenopausal women substituted with 17 beta-estradiol and norethisterone acetate. A two-year follow-up study. Hypertension 1995; 25:848-853. [ Links ]
46. Baylis C, Vallance P. Measurement of nitrite and nitrate levels in plasma and urine--what does this measure tell us about the activity of the endogenous nitric oxide system? Curr Opin Nephrol Hypertens 1998; 7: 59-62. [ Links ]
47. Miyamoto Y, Saito Y, Nakayama M, Shimasaki Y, Yoshimura T, Yoshimura M, et al. Replication protein A1 reduces transcription of the endothelial nitric oxide synthase gene containing a -786T-->C mutation associated with coronary spastic angina. Hum Mol Genet 2000; 9: 2629-2637. [ Links ]
48. Jeerooburkhan N, Jones LC, Bujac S, Cooper JA, Miller GJ, Vallance P, et al. Genetic and environmental determinants of plasma nitrogen oxides and risk of ischemic heart disease. Hypertension 2001; 38: 1054-1061. [ Links ]
49. Nasreen S, Nabika T, Shibata H, Moriyama H, Yamashita K, Masuda J, et al. T-786C polymorphism in endothelial NO synthase gene affects cerebral circulation in smokers: possible gene-environmental interaction. Arterioscler Thromb Vasc Biol 2002; 22: 605-610. [ Links ]
50. Nagassaki S, Metzger IF, Souza-Costa DC, Marroni AS, Uzuelli JA, Tanus-Santos JE. eNOS genotype is without effect on circulating nitrite/nitrate level in healthy male population. Thromb Res 2005; 115: 375-379. [ Links ]
51. Wang XL, Mahaney MC, Sim AS, Wang J, Wang J, Blangero J, et al. Genetic contribution of the endothelial constitutive nitric oxide synthase gene to plasma nitric oxide levels. Arterioscler Thromb Vasc Biol 1997; 17: 3147-3153. [ Links ]
52. Tsukada T, Yokoyama K, Arai T, Takemoto F, Hara S, Yamada A, et al. Evidence of association of the ecNOS gene polymorphism with plasma NO metabolite levels in humans. Biochem Biophys Res Commun 1998; 245:190-193. [ Links ]
53. Wang XL, Sim AS, Badenhop RF, McCredie RM, Wilcken DE. A smoking-dependent risk of coronary artery disease associated with a polymorphism of the endothelial nitric oxide synthase gene. Nat Med 1996; 2: 41-45. [ Links ]
54. Wang XL, Sim AS, Wang MX, Murrell GA, Trudinger B, Wang J. Genotype dependent and cigarette specific effects on endothelial nitric oxide synthase gene expression and enzyme activity. FEBS Lett 2000; 471: 45-50. [ Links ]
55. Komatsu M, Kawagishi T, Emoto M, Shoji T, Yamada A, Sato K, et al. ecNOS gene polymorphism is associated with endothelium-dependent vasodilation in Type 2 diabetes. Am J Physiol Heart Circ Physiol 2002; 283: H557-561. [ Links ]
56. Senthil D, Raveendran M, Shen YH, Utama B, Dudley D, Wang J, et al. Genotype-dependent expression of endothelial nitric oxide synthase (eNOS) and its regulatory proteins in cultured endothelial cells. DNA Cell Biol 2005; 24: 218-224. [ Links ]
57. Dipple KM, McCabe ER. Phenotypes of patients with "simple" Mendelian disorders are complex traits: thresholds, modifiers, and systems dynamics. Am J Hum Genet 2000; 66: 1729-1735. [ Links ]
58. International HapMap Project. En: URL: http://www.hapmap.org/. [ Links ]
59. Goldstein DB, Ahmadi KR, Weale ME, Wood NW. Genome scans and candidate gene approaches in the study of common diseases and variable drug responses. Trends Genet 2003; 19: 615-622. [ Links ]
60. Weale ME, Depondt C, Macdonald SJ, Smith A, Lai PS, Shorvon SD, et al. Selection and evaluation of tagging SNP in the neuronal-sodium-channel gene SCN1A: implications for linkage-disequilibrium gene mapping. Am J Hum Genet 2003; 73: 551-565. [ Links ]
61. Ahmadi KR, Weale ME, Xue ZY, Soranzo N, Yarnall DP, Briley JD, et al.A single-nucleotide polymorphism tagging set for human drug metabolism and transport. Nat Genet 2005; 37: 84-89. [ Links ]
62. Pardi F, Lewis CM, Whittaker JC. SNP selection for association studies: maximizing power across SNP choice and study size. Ann Hum Genet 2005; 69: 733-746. [ Links ]

