











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink1. INTRODUCCIÓN
La transformación genética de microorganismos en el laboratorio es una técnica rutinaria de enorme utilidad tanto para trabajos en ingeniería genética como estudios de genética básica y aplicada. [1,2]. Este procedimiento fue descrito por primera vez en Escherichia coli por Mandel y Higa [1]. A partir de esa fecha, la eficiencia del protocolo se ha ido mejorando sustancialmente prolongando la exposición de las células al CaCl2 o sustituyéndolo por otros cationes (Rb+, Mg2+ Mn2 + o K+) y por la adición de otros compuestos que permiten introducir cualquier plásmido en su forma circular o súperenrollada en casi todo tipo de bacteria [3].
En la naturaleza, solo muy pocas especies bacterianas tienen capacidad de captar ADN foráneo de forma natural mediante la transformación, transducción o conjugación de genes [4]; y hay pocos estudios que describan como sucede de manera natural la transformación de las bacterias y cómo experimentalmente los transposones, integrones o casetes de genes se mueven horizontalmente [5]. Sin embargo, a nivel de laboratorio se puede inducir de forma artificial; teniendo en cuenta dos parámetros principales que están implicados en la transformación eficiente de una bacteria, el primero es el método utilizado para inducir la competencia para la transformación necesario para la adopción de material genético externo y el segundo es la constitución genética de la cepa que se está transformando [6]. Existen varios métodos para la transformación artificial bacteriana como la conjugación, que requiere de contacto físico entre el receptor y el donante que contiene el plásmido conjugativo [4], las micro ondas de choque [7], los tratamientos a base de CaCl2 [8] y la electroporación [9,10]; estos dos últimos son los más utilizados.
A nivel de laboratorio algunas cepas bacterianas de Escherichia coli son las más utilizadas, gracias al vasto conocimiento que se tiene acerca de su genética, bioquímica y biología molecular, siendo generalmente el primer sistema que se escoge para la expresión de proteínas recombinantes en bacterias [1]. También estas son empleadas para múltiples actividades, que van desde la conservación y propagación del ADN plasmídico de clones hasta la creación de grandes bibliotecas para la determinación de la secuencia de un genoma [11]. Mientras que para trabajos de expresión de proteínas recombinantes en plantas, el sistema de transformación vegetal más empleado es vía Agrobacterium tumefaciens. Una bacteria típica del suelo, gram negativa, la cual representa una situación única en la naturaleza, por tener la capacidad de transferir el elemento genético T-DNA, a un organismo eucariota superior, con su subsiguiente integración y expresión en el genoma del hospedero [12,13]. Gracias a la facilidad que presenta esta bacteria para transferir material genético, varias especies vegetales de importancia agronómica son rutinariamente transformadas usando A. tumefaciens; la lista de especies que son susceptibles a transformación mediada por A. tumefaciens crece año tras año [14].
En el laboratorio de Genética Molécular Vegetal de Corpoica Tibaitatá se cuenta con varios ADN desnudos que presentan genes reporteros gus y gfp, y en esta condición permite que se aumente las posibilidades de degradación y se dificulta su utilización para transformación genética de plantas. Aunque se han reportado varios métodos para la transformación artificial de bacterias no todas las cepas transforman con las mismas condiciones, por lo tanto el objetivo de este trabajo fué estandarizar protocolos de transformación genética en E. coli DH5a y A. tumefaciens como LBA 4404, EHA 105 y C58 para la generación de una colección de constructos génicos de manera rápida y confiable. Con una colección base de constructos génicos en cepas bacterianas se logra una conservación estable del ADN, replicación rápida del plásmido y su disponibilidad inmediata para procesos de transformación en plantas.
2. METODOLOGÍA
Esta investigación se realizó en el laboratorio de Genética Molécular Vegetal, de la Corporación Colombiana de Investigación Agropecuaria (CORPOICA-Tibaitatá), ubicada en el municipio de Mosquera, Cundinamarca.
2.1 Material biológico
La cepa de E. coli DH5a y A. tumefaciens cepas LBA4404, EHA105 y C58 fueron cedidas por el laboratorio de Biología Molecular del Departamento de Biología de la Universidad Nacional de Colombia. La cepa DH5a de E. coli fue crecida durante 48 horas a 37 °C, mientras que las cepas de Agrobacterium se crecieron durante 48 horas a 28°C y mantenidas en medio Luria Bertani (LB), suplementado con estreptomicina 50 mg/L para la cepa LBA4404 y rifampicina 50 mg/L para las cepas EHA105 y C58.
2.2 Material genético
Se emplearon 23 vectores binarios pertenecientes a la serie pCAMBIA, el vector pSK1019 donado por el Dr. Seogchan Kang de la Universidad de Pensilvania y el vector pMP2482 cedido por la Dra. Nicolette Quaedulig del Institute of Molécular Plant Sciences (Holanda), para un total de 25 vectores (Tabla 1).

2.3 Transformación bacteriana
La bacteria E. coli cepa DH5a se transformó por choque térmico empleando el protocolo descrito por Pope y Kent [8] con algunas modificaciones como la disminución de la cantidad de ADN plasmídico a 50 ng/µl y del tiempo de choque térmico a 45 segundos.
Para transformar las cepas desarmadas de A. tumefaciens LBA4404, EHA105 y C58 se probaron 2 métodos: choque térmico y electroporación. Para la primera, se empleó el protocolo descrito en el manual de biología molécular de plantas [15] y para la segunda, que se utilizó el electroporador Biorad Gene Pulser®, se evaluaron dos protocolos de preparación y electroporación de células de A. tumefaciens [15,16]. Walkerpeach and Velten [15] emplean HEPES pH 7, para los lavados y resuspensión de las células, con un voltaje de electroporación de 2.5 KV y Utermark and Kar-lovsky (16) emplean agua grado molécular en la preparación de la solución bacteriana y un voltaje de electroporación de 1.8 KV. Adicionalmente se evaluaron los voltajes 1.5, 2.0 y 2.2 KV. Como control se sembró sobre el medio de selección, 100 (iL de células bacterianas sometidas al proceso de transformación, pero omitiendo el paso de adicionar ADN plasmídico; las cepas de A. tumefaciens se mantuvieron antes de utilizarlas, en un baño de hielo al igual las cubetas de electroporación y los tubos eppendorf de 1.5 mL estériles.
2.4 Extracción y visualización del ADN plasmídico.
Para los ADN plasmídicos que se encontraban en las cepas de E. coli se empleó el protocolo de mini-prep de lisis alcalina, reportado por Sambrook and Russell [2]; y se visualizó en geles de agarosa al 1% teñidos con bromuro de etidio a 0.1 µg/mL usando el software Gene-snap. La concentración y pureza del ADN se determinó utilizando un espectrofotómetro UV 260/280 (Beckman Du530).
Con respecto a los ADN plasmídicos que se encontraban en las cepas de A. tumefaciens se realizaron 2 métodos de extracción: la mini-prep de lisis alcalina [2] y la extracción de ADN crudo descrita por Hernández et al [17] con la modificación de la solución donde se resuspende la bacteria que fue 50 µL de TEE (1% de triton X 100, 20 mM de tris-HCl, pH 8.5 y 1 mM de EDTA, pH 8,0), el resto del procedimiento se mantuvo (tomar media colonia bacteriana transformada y suspenderla en una solución para luego incubarla por 10 minutos a 95°C, transcurrido el tiempo se centrifuga por 5 minutos a 7000 rpm); el sobrenadante es usado como ADN base en la reacción de PCR.
2.5 Confirmación de transformación con los vectores pSK1019, pMP2482 y pCAMBIA.
La confirmación de transformación con los vectores empleados se hizo de dos maneras, para los plásmidos pSK1019 y pMP2482 mediante PCR verificando la presencia del gen y para los pCAMBIA a través de cortes con enzimas de restricción para para verificar el tamaño de estos.
Al plásmido pSK1019 que contiene el gen egfp se le realizó PCR con los iniciadores glGFP-R 5´-GCCGAGCTCAG ATCTCACTTGTACAGCTC GTCCATGCC-3´ y glGFP-F 5´GCCGGAATTCATGAGCAAGGGCGAGGAACTGTTC3´ plasmídico reportados por Abello et al et al [18]. Las condiciones de la reacción fueron las siguientes: 100 ng de ADN plasmídico, 0.3 µM de cada iniciador, 0.2 mM de dNTPs, 1X de buffer de PCR, 1.5 mM de MgCl2 y 1 unidad de Taq polimerasa. La amplificación fue realizada en un termociclador (PT100 MJ Research) usando un programa de 35 ciclos de 95 o C por 1 min, 60 o C por 1 min, 72 oC por 1 min, con una extensión final de 72 o C por 3 min.
El plásmido pMP2482 que contiene el gen Npt ll, se confirmó con los iniciadores 5'-CCACCAT-GATATTCGGCAAC-3'y 5'- GTGGAGAGGCTA-TTCGGC TA3', reportados por Saíni and Jaiwal [19]; las condiciones de reacción fueron: 2 µL de ADN total crudo, 0.3 µM de cada iniciador, 0.2 mM de dNTPs, buffer de PCR 1X, 1.5 mM de MgCl2 y 1 unidad de taq polimerasa. El proceso de amplificación de los iniciadores consistió en 38 ciclos de 94 oC por 1 min, 62 oC por 1 min, 72 oC por 1 min y una extensión final de 72 oC por 7 min.
En cuanto a los 23 vectores pCAMBIA, la verificación se realizó mediante cortes con la enzima de restricción EcoR 1 y Xho 1 que presentan uno y dos sitios de corte respectivamente. (Promega).
3. RESULTADOS Y DISCUSIÓN
3.1 Transformación de E. coli.
Se observó en el medio de selección, el crecimiento de unidades formadoras de colonias (UFC) bien definidas, con bordes lisos y apariencia cremosa con un promedio de 105 UFC/100 lL. En las tres réplicas establecidas como control no se observaron crecimiento. Este protocolo permitió la obtención de colonias transformadas, con una eficiencia de transformación de 5.2 x 103 UFC/20 ig de ADN plasmídico, aunque esta eficiencia no es tan alta como la reportada por Sambrook and Russell [2] y Pope and Kent [8], donde alcanzan eficiencias de transformación de ~5 * 106 UFC/ µg de ADN plasmídico, ella es suficiente para obtener transformantes en menor tiempo y menos cantidad de ADN cuando se trabaja de manera masiva.
3.2 Confirmación de vectores transformados en Escherichia coli.
A partir de las colonias obtenidas sobre los medios de selección se realizó la extracción del ADN plasmídico y se cuantificó por espectrofotometría hecho que mostró concentraciones promedio de 1550 (µg/mL. Este ADN fue empleado para la confirmación por PCR del vector pSK1019 hecho que reveló un fragmento de 756 pb (Figura 1) que coincide con lo reportado por Abello et al [18] y del vector pMP482 con un fragmento de 540 pb que concuerda con lo reportado por Saíni and Jaiwal [19]. (dato no mostrado).
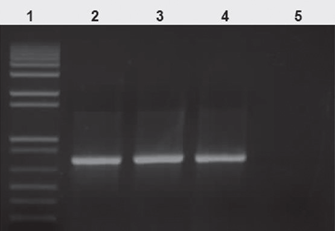
Figura 1 Perfil electroforético de la PCR, con los iniciadores egfp, para el vector pSK1019, transformado en E. coli cepa DH5α. Carril 1, marcador de peso de 1 Kb plus. Carril 2, PCR con DNA de la colonia 1. Carril 3, PCR con DNA de la colonia 2. Carril 4, PCR con DNA de la colonia 3. Carril 5, control absoluto (agua).
Los 23 vectores pCAMBIA se confirmaron por cortes con enzimas de restricción y se obtuvieron los pesos moléculares esperados según el mapa de restricción de cada vector. Como la enzima Eco RI presenta un único punto de corte se obtuvo su linealización y se observó una banda correspondiente al peso de cada vector (Figura 2) , mientras que la enzima Xho I que presenta dos puntos de corte, se observó una banda de 1094 pb que corresponde al corte del gen de la higromicina y otra de mayor tamaño la cual varía dependiendo del peso de cada vector, como se muestra en la figura 3.

Figura 2 Perfil electroforético del corte de restricción del DNA plasmídico de E. coli cepa DH5α con la enzima Eco RI. Carril 1 y 13, marcador de peso de 1 Kb plus. Carril 2, pC1304. Carril 3, pC1381. Carril 4, pC1381Xa. Carril 5, pC1381Xb. Carril 6, pC1381Xc. Carril 7, pC1391. Carril 8, pC1391Xa. Carril 9, pC1391Xb. Carril 10, pC1391Xc. Carril 11, pC1381Z. Carril 12, pC1391Z.

Figura 3 Perfil electroforético del corte de restricción del DNA plasmídico de E. coli cepa DH5α con la enzima Xho I. Carril 1, marcador de peso de 1 Kb plus. Carril 2, pC1300. Carril 3, pC1380. Carril 4, pC1390. Carril 5, pC1305.1. Carril 6, pC2300. Carril 7, pC1301. Carril 8, pC2301. Carril 9, pC13021. Carril 10, pC1303. Carril 11, pC1305.1.
3.3 Transformación de Agrobacterium tumefaciens
La transformación de las cepas de A. tumefaciens de este trabajo con choque térmico no fue efectiva pues no se logró obtener crecimiento de colonias transformantes; López [20] obtuvo una baja eficiencia, promedio de recuento de 0,6 ufc/100 µL, cuando también utilizaron este método. Lo anterior se podría explicar por la estructura que presenta la pared celular de estas bacterias, que contienen tres capas de la membrana externa, una membrana citoplasmática y un espacio periplásmico entre ellas, que varía en su grosor desde varias unidades hasta cientos de nanómetros, a diferencia de la pared celular de las demás bacterias gram negativas contienen solo una membrana externa muy asimétrica [20]. Por lo tanto se hace difícil labilizar la pared y por ende crear poros que permitan la entrada del ADN plasmídico para obtener células transformadas.
No todas las bacterias se comportan de igual manera para los diferentes métodos de transformación como lo demuestran en 1990, Diver et al [10] al transformar la bacteria gram negativa Pseudomonas aeruginosa PAO1 con el plásmido pLAFR1 a través de electrotransformación y transformación química convencional con MgCl2. Con el primer método obtuvieron un aumento de 1500 veces en la eficiencia de transformación, y además demostraron que el voltaje aplicado y la composición de tampón de electroporación tienen el mayor efecto sobre la eficiencia de la transformación. También es importante mencionar que la constitución genética de la cepa a transformar puede influir dramáticamente en el resultado de los experimentos de transformación [6]. Por ello es necesario identificar el método de transformación para la bacteria a utilizar.
Al realizar el método de electroporación, los mejores resultados se obtuvieron empleando el protocolo descrito por Utermark and Karlovsky [16], con la modificación del voltaje que fue de 2.0 kV., obteniéndose una eficiencia de transformación de 3 x 103 UFC/20 µg de ADN basado en el número de unidades formadoras de colonias obtenidas por cada 200 µl de células bacterianas sembradas sobre medio de selección.
En 2008, Utermark and Karlovsky [16] reportan que concentraciones de 0.1 ng/µL de ADN plasmídico son suficientes para obtener transformantes de A. tumefaciens, sin embargo en este estudio empleando este protocolo, solo se obtuvo crecimiento de colonias transformantes, a una concentración mínima de 10 ng/µL de ADN plasmídico en un volumen de 20 µL de células electrocompetentes y voltaje de 2.0 kV. Esto podría verse explicado, que durante el proceso de extracción quedaran impurezas, lo que podría conllevar a sobreestimaciones en el momento de la cuantificación del ADN. Las colonias transformantes se observaron luego de plaquear 200 µL de células bacterianas sometidas al proceso de electroporación e incubadas por 48 hr a 28°C sobre medio de selección.
3.4 Extracción de ADN plasmídico y confirmación de vectores transformados en A. tumefaciens
La extracción de ADN plasmídico crudo utilizada para A. tumefaciens cepas LBA 4404, EHA 105 y C58 que contenían los vectores pSK1019 y pMP2482 muestra ser eficiente al confirmar por PCR los vectores pSK1019 (Figura 4) y pMP2482 (dato no mostrado) pues se encontró que para ambos vectores, el 100% de colonias evaluadas fueron positivas. Para las bacterias con los vectores pCAMBIA también se obtuvieron eficientes resultados con el protocolo mini-prep de lisis alcalina, aunque es más largo y dispendioso. Estos resultados demuestran que el empleo de ADN crudo es confiable para la detección rápida por PCR de transformantes bacterianos, permitiendo de esta forma ahorrar tiempo y dinero en los laboratorios donde los procesos de transformación bacteriana se hacen de forma masiva y rutinaria. Por cada vector, se confirmaron tres colonias transformantes tomadas al azar.

Figura 4 Perfil electroforético de la PCR, con los iniciadores npt ll para el vector pMP2482, transformado en A. tumefaciens cepa EHA 105, LBA 4404 y C58. Carril 1, marcador de peso 1 Kb plus. Carril 2, PCR con ADN crudo de cepa EHA 105. Carril 3, PCR con ADN crudo de cepa LBA 4404. Carril 4, PCR con ADN crudo de cepa C58. Carril 5, Control positivo con ADN crudo de cultivo fresco de pMP2482 cepa E. coli. Carril 6. Control positivo con ADN crudo cultivo en glicerol de pMP2482 cepa E. coli. Carril 7, Control con agua.
CONCLUSIONES
Para transformar la cepa Escherichia coli DH5α mediante choque térmico se puede utilizar 45 segundos y 50 ng de ADN plasmídico para obtener de manera rápida y confiable transformantes cuando se trabaja transformación genética de manera masiva y rutinaria.
Las cepas de Agrobacterium tumefaciens LBA 4404, EHA 105 y C 58 como se pudo comprobar en este estudio no se pudieron transformar con el método de choque térmico, por ello se realizó la estandarización a través de electroporación siendo esta efectiva.
El empleo de ADN crudo es confiable para la detección rápida por PCR de transformantes bacterianos, permitiendo de esta forma ahorrar tiempo y dinero en los laboratorios.
Es necesario estandarizar los protocolos de transformación en las cepas bacterianas a utilizar porque no todas transforman con las mismas condiciones y este trabajo puede ser un referente para la estandarización en otros laboratorios de transformacion genética cuando se trabaja de manera masiva.

