











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La retinopatía de Purtscher fue descrita en 1910 por Omar Purtscher en un paciente masculino, posteriormente a un trauma craneoencefálico severo1. Es una enfermedad ocular rara, asociada con una patología sistémica que se mani fiesta con áreas de blanqueamiento intrarretiniano y zonas de aclaramiento a cada lado de las arteriolas, vénulas y arteriolas precapilares de la retina. Esto contrasta con las manchas de algodón, que tienen bordes mal definidos y se encuentran superficialmente sobre los vasos1,2. A escala mundial, se ha descrito una incidencia de la enfermedad de aproximadamente 0,24 casos por millón de personas por año1.
La fisiopatología de esta entidad todavía no es clara, se sugiere una asociación con un componente microvasculopático embólico oclusivo, con pérdida súbita de la visión luego de trauma o al inicio de enfermedades sistêmicas asociadas como el lupus eritematoso sistémico (LES) (3.
El LES es una enfermedad inflamatoria autoinmune multisistémica en la cual el compromiso ocular puede ser una de las primeras manifestaciones, con una incidencia del 29% en pacientes con enfermedad activa4.
El diagnóstico de la retinopatía Purtscher-like es esencial mente clínico, basado en el compromiso de la agudeza visual o de los campos visuales, tras el inicio de una patología sub yacente asociada2.
En el examen oftalmológico del fondo de ojo típicamente se encuentran manchas algodonosas (Purtscher-flecken), además de hemorragias retinianas5. Las ayudas diagnósticas com plementarias incluyen la tomografía de coherencia óptica (OCT), la angiografía por tomografía de coherencia óptica (angio-OCT) y la angiografía fluoresceínica (AF), las cuales pro porcionan información fundamental para el diagnóstico y el seguimiento de los pacientes5.
El tratamiento de esta patología aún no es claro, se ha evidenciado que existe mejoría sintomática con el uso de corticosteroides, pero hay revisiones que no son concluyentes y se basan en la opinión de expertos. Se presenta el caso de un paciente con retinopatía de Purtscher-like como primera manifestación de LES.
Descripción del caso
Paciente masculino de 25 años con cuadro de 10 meses de evolución de hipoacusia aislada y alopecia progresiva. Ocho meses después inicia con episodios de cefalea global, asociado con picos febriles objetivos diarios de hasta 40° C, con apari ción de adenomegalias cervicales no dolorosas, acompañado de astenia, adinamia, mialgias y parestesias en miembros inferiores, además de pérdida objetiva de peso de aproxima damente 10 kg en dos meses.
Un mes después inicia con visión borrosa, por lo cual con sulta al Servicio de Oftalmología de una clínica privada de la ciudad de Medellín, Colombia. En el examen oftalmológico ini cial se encontraba con una agudeza visual en ojo derecho de 20/25 y en ojo izquierdo de 20/20, sin alteraciones en anexos oculares ni en segmento anterior, y con una presión intraocu lar normal en ambos ojos. En el fondo de ojo se observaba la presencia de manchas algodonosas (Purtscher-flecken) desde el nervio óptico hacia la mácula en ambos ojos. En el ojo derecho presentaba una hemorragia en mancha sobre la arcada tem poral superior, con dilatación vascular marcada a nivel inferior (fig. 1).
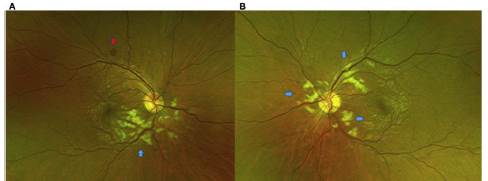
Figura 1 A) Foto a color del ojo derecho: nervio con excavación de 0,1, de bordes definidos, vasos con dilatación de predominio temporal inferior, con rectificación a nivel macular, manchas blancoalgodonosas peripapilares de predominio inferior, hipopigmentación de la retina a nivel peripapilar respetando los vasos, correspondiente a Purtscher-flecken (flecha azul). Hemorragia en mancha hacia la arcada temporal superior (flecha roja). B) Foto a color ojo izquierdo: nervio con excavación de 0,1, de bordes definidos, vasos dilatados de predominio temporal superior, manchas blancoalgodonosas peripapilares, hipopigmentación de la retina a nivel peripapilar, respetando los vasos, correspondiente a Purtscher-flecken (flechas azules).
Se solicita OCT macular (fig. 2) y Angio-OCT de ambos ojos (fig. 3), las cuales muestran una pérdida parcial del con torno foveal en el ojo derecho, presencia de lesiones quísticas intrarretinianas a predominio del sector inferonasal con com promiso foveal, engrosamiento y aumento de la reflectividad de los segmentos internos del sector nasal (retinitis vs. congestión vascular), con grosor coroideo levemente aumentado, pero con coriocapilaris congestiva y dilatación de los vasos de Haller. En el ojo izquierdo se observa pérdida parcial del contorno foveal, presencia de lesiones quísticas intrarretinianas, así como engrosamiento y aumento de la reflectividad de los segmentos internos del sector nasal (retinitis vs. con gestión vascular), con grosor coroideo levemente aumentado, coriocapilaris congestiva y dilatación de los vasos de Haller. El angio-OCT complementario sugiere una oclusión vascular inferotemporal con áreas de no perfusión en ojo derecho y se recomienda complementar estudios con angiografía fluores ceínica de ambos ojos (fig. 4).
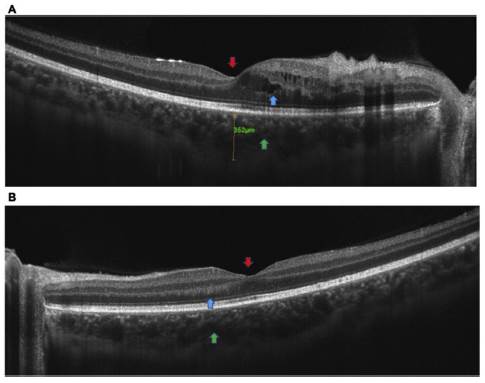
Figura 2 A) OCT ojo derecho: pérdida parcial del contorno foveal (flecha roja), presencia de lesiones quísticas intrarretinianas a predominio del sector inferonasal con compromiso foveal (flecha azul), engrosamiento y aumento de la reflectividad de los segmentos internos del sector nasal (congestión vascular), con grosor coroideo levemente aumentado, pero con coriocapilaris congestiva y dilatación de los vasos de Haller (flecha verde). B) OCT ojo izquierdo: pérdida parcial del contorno foveal (flecha roja), con engrosamiento y aumento de la reflectividad de los segmentos internos del sector nasal (flecha azul), con grosor coroideo levemente aumentado, coriocapilaris congestiva y dilatación de los vasos de Haller (flecha verde).
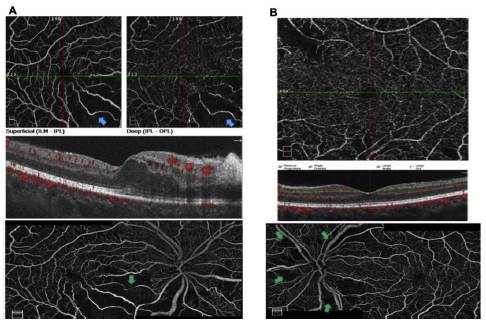
Figura 3 A) Angio-OCT ojo derecho: se observan áreas que sugieren hipoperfusión a nivel macular inferonasal, tanto en el plexo superficial como en el profundo, que se correlacionan con las áreas de edema macular (flechas azules). Reconstrucción OCT-A en la que se observa dilatación vascular peripapilar, zonas que sugieren hipoperfusión a nivel peripapilar nasal y temporal, como también a nivel macular inferonasal (flecha verde). B) Angio-OCT ojo izquierdo: plexo profundo con zonas de posible hipoperfusión superotemporal vs. artefactos, explicados por lesiones blanquecinas. Reconstrucción de angio OCT-A de nervio y mácula que muestra dilatación vascular de predominio superior, con zonas de no perfusión peripapilar (flechas verdes).
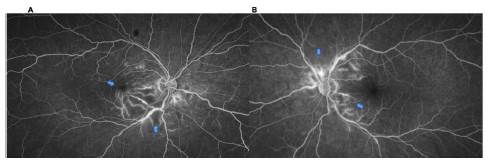
Figura 4 Angiografía fluoresceínica de ambos ojos. A) Ojo derecho: imagen en fases tardías en la que se comienzan a observar hiperfluorescencia a nivel venoso de la arcada temporal inferior y vasos maculares a nivel nasal, con zonas de hipoperfusión macular inferonasal y peripapilar inferonasal (flechas azules). B) Ojo izquierdo: imagen de angiografía en la cual se observa hipoperfusión peripapilar a nivel nasal superior, temporal superior e inferior, con dilatación vascular venosa a nivel superior, además de tinción perivascular venosa tardía (flechas azules).
Dentro de los paraclínicos iniciales realizados se encuentra un perfil infeccioso negativo (toxoplasmosis, citomegalovirus, sífilis, hepatitis C, hepatitis B, virus de inmunodeficiencia humana y dengue), test de tuberculina negativo en 0 (cero) y tomografía axial computarizada (TC) simple de tórax negativa. El paciente presenta un extendido de sangre periférico, per fil renal y hepático normal, sin embargo, llaman la atención unos niveles de lactato deshidrogenasa elevados, asociados con anticuerpos antinucleares positivos con patrón moteado de 1:160, anticuerpos anti-Smith (Sm) positivos, hipocomplementemia C3 y linfopenia, sin otras citopenias asociadas. Además, presenta una prueba de Coombs directo positivo, sin evidencia de hemólisis. Se decide hospitalizar para evaluación y estudios.
Con posterioridad, se lleva a cabo una resonancia mag nética nuclear contrastada de cráneo, la cual muestra atrofia cortical y escasas hiperintensidades periventriculares difusas, sin signos de patología desmielinizante. La punción lumbar con líquido cefalorraquídeo (LCR) se encuentra dentro de los límites normales, con tinta china y KOH en LCR negativo. La TC de cuello contrastada muestra pequeños ganglios de 5 mm en las estaciones II y V, sin conglomerado de adenopatías. En la TC de tórax contrastada se observan pequeños ganglios de localización paratraqueal derecha menores de 5 mm, en tanto que en la TC de abdomen contrastada se encuen tran ganglios inguinales bilaterales de 5 mm en su eje corto. La electromiografía de miembros inferiores presenta denervación aislada tibial anterior izquierdo por compromiso de nervio fibular.
Adicionalmente, se realizan estudios para descartar sín drome antifosfolípido, con anticuerpos anticardiolipina IgM e IgG negativos, beta 2 glicoproteína 1 IgM e IgG negativos y anticoagulante lúpico negativo. El reporte de validación de anticuerpos antifosfolípidos IgM glicoproteína I, cardiolipina, fosfatidil serina, fosfatidil inositol y ácido fosfatídico).
Resultados
Con todo lo anterior y teniendo en cuenta los criterios revi sados de la Liga Europea Contra el Reumatismo (EULAR) y el Colegio Americano de Reumatología (ACR), se realiza un diagnóstico de LES de novo y se inicia manejo con pulsos de metilprednisolona 500 mg, intravenosos (IV), cada 24 horas, por tres dosis, continuando 50 mg, cada 24 horas, vía oral, más cloroquina 150 mg, cada 24 horas, vía oral6.
Posteriormente a los pulsos de esteroide intravenoso (IV), el paciente refiere mejoría de sus síntomas visuales, sin embargo, en evaluaciones oftalmológicas posteriores se encuentran cambios hemorrágicos leves también en el ojo izquierdo, con una agudeza visual en ambos ojos de 20/20 (fig. 5). Se decide, en consecuencia, iniciar bolos de ciclofosfamida de 750 mg, IV.
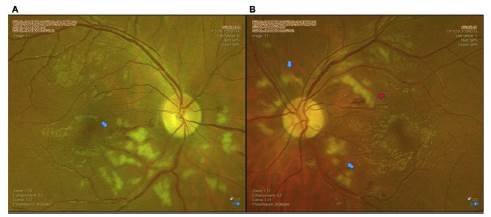
Figura 5 Fotografías a color de ambos ojos de control una semana después de iniciado el tratamiento. A) Ojo derecho: persisten vasos con dilatación de predominio temporal inferior, con rectificación a nivel macular, manchas blancoalgodonosas peripapilares de predominio inferior e hipopigmentación de la retina a nivel peripapilar respetando los vasos, correspondiente a Purtscher-flecken (flecha azul). B) Ojo izquierdo con cambios hemorrágicos leves (flecha roja). Persisten vasos dilatados de predominio temporal superior, manchas blancoalgodonosas peripapilares, hipopigmentación de la retina a nivel peripapilar, respetando los vasos, correspondiente a Purtscher-flecken (flechas azules).
Se hace seguimiento al paciente y se observa una marcada mejoría en los hallazgos retinianos luego de tres meses del ini cio de la terapia instaurada (corticosteroides y ciclofosfamida IV mensual), con OCT de mácula de control con evidencia de disminución del edema a nivel nasal en el ojo derecho, y en el examen oftalmológico se encuentra agudeza visual en el ojo derecho con estenopeico de 20/20 (previa de 20/25) y en el ojo izquierdo con estenopeico de 20/20-2 (previa de 20/20), sin alte raciones en los anexos oculares ni en el segmento anterior y presión intraocular dentro de límites normales en ambos ojos (fig. 6). Al evaluar el fondo de ojo, se encuentran manchas algo donosas leves y hemorragias puntiformes a nivel peripapilar inferior en el ojo derecho, con mácula sana y retina periférica sana, mientras que en el ojo izquierdo se observan manchas algodonosas leves en área inferotemporal, con mácula y retina periférica sanas.
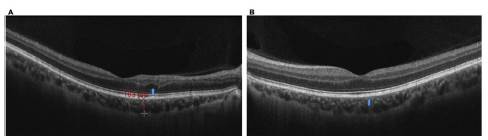
Figura 6 OCT de mácula ambos ojos: imágenes de control a los tres meses de seguimiento. A) Ojo derecho: mácula con alteración de la continuidad lineal de las capas del plexo retinal externo yuxtafoveal, sin compromiso de la zona elipsoide, con reflectividad alta del complejo EPR/coriocapilaris con mínimos cambios atróficos (flecha azul). B) Ojo izquierdo: mácula con arquitectura normal, con reflectividad alta del complejo EPR/coriocapilaris y mínimos cambios atróficos (flecha azul).
Discusión
La retinopatía Purtscher-like es una entidad rara que cursa con severa angiopatía, se caracteriza por lesiones confluen tes de manchas algodonosas en el polo posterior, asociado con hemorragias retinianas, con disminución súbita de la agudeza visual en el contexto de una enfermedad sistémica, en este caso el LES5.
El mecanismo exacto de esta entidad sigue sin estar claro, la evidencia actual sugiere una asociación con un compo nente microvasculopático embólico oclusivo, principalmente de arteriolas precapilares, con pérdida súbita de la visión luego de traumas o al inicio de enfermedades sistémicas especiales. Se han descrito hallazgos retinianos similares en un amplio rango de patologías como pancreatitis, falla renal y enferme dades autoinmunes como LES, entre otras7,8.
En pacientes con diagnóstico de LES, la retinopatía suele manifestarse en aquellos con enfermedad activa y se asocia comúnmente con síndrome antifosfolípido. La presencia de anticuerpos antifosfolípido aislados o en asociación con LES incrementa el riesgo de retinopatía9.
El diagnóstico de la retinopatía Purtscher-like es clínico, con una presentación amplia que va desde pérdida súbita de la visión en horas o días hasta pérdida del campo visual cen tral, paracentral o de escotomas arqueados, usualmente con visión periférica preservada, tras el inicio de una patología subyacente asociada2.
Los signos encontrados en el fondo de ojo incluyen manchas algodonosas (Purtscher-flecken), generalmente bila terales, además de hemorragias retinales que han sido descritas hasta en el 83-92% de los casos5.
Wu et al. informaron una serie de casos de retinopatía Purtscher-like en pacientes previamente diagnosticados con LES entre el 2002 y el 2013. En el fondo de ojo encontraron lesiones Purtscher-flecken, manchas de algodón, hemorragias retinianas, edema macular, edema del disco óptico y una man cha roja pseudocereza. Las anormalidades de la angiografía con fluoresceína incluyeron áreas de no perfusión capilar, fuga tardía, tinción peripapilar, oclusión precapilar y llenado lento de los vasos10.
Li et al. describieron en la angio-OCT áreas maculares de no perfusión extensas, con compromiso de plexos capilares internos y profundos, OCT con edema macular y AF con áreas de no perfusión, isquemia retinal y llenado lento de los vasos. Algunos estudios recientes han demostrado edema macular cistoide y fluido subretinal asociado11. En nuestro caso, la OCT y la angiografía evidenciaron edema macular cistoide leve, con pérdida parcial del contorno foveal, grosor coroideo levemente aumentado, coriocapilaris congestiva, dilatación de los vasos de Haller y oclusión vascular inferotemporal con áreas de no perfusión.
Los diagnósticos diferenciales que es preciso tener en cuenta incluyen oclusión de rama o de arteria central de la retina, conmoción reque es preciso tiniana y embolismo graso. También debe descartarse la asociación con otras enfermeda des sistémicas como la retinopatía por VIH, que también cursa con manchas algodonosas2.
En la actualidad, en la literatura no existen guías clínicas que orienten el tratamiento de estos pacientes, por lo cual el manejo se ha basado en la opinión de expertos, en la que se observan dos estrategias o líneas de manejo: administra ción de corticosteroides o inmunosupresores vs. seguimiento sin tratamiento farmacológico, pues se considera que el curso de la enfermedad tiende a ser autolimitado. Se ha mos trado mejoría significativa con el uso de corticosteroides, pero hay revisiones que no son concluyentes, ya que se requiere una homogenización de los pacientes y de las patologías desencadenantes2,12.
Nuestro reporte de caso ilustra la historia natural de la retinopatía Purtscher-like asociada con enfermedad sistémica tipo LES, la cual por su presentación clínica, manifestaciones y hallazgos oftalmológicos hizo pensar en esta patología, con mejoría de signos y síntomas luego del inicio de corticosteroides y bolos de ciclofosfamida intravenosa mensual9.
Conclusiones
Se trata de un caso de retinopatía de Purtscher-like con vasculitis bilateral de tipo oclusiva, cuyo compromiso ocular y valoración oftalmológica temprana orientaron e hicieron posi ble el diagnóstico de LES de novo, como también el inicio de tratamiento de forma oportuna en un paciente joven.
Consideramos fundamental la valoración oftalmológica completa en pacientes con sospecha de enfermedades autoinmunes, ya que ciertos hallazgos oftalmológicos son la primera manifestación de algunas enfermedades sistémicas, lo que nos puede llevar a realizar diagnósticos tempranos e integrales con el fin de mejorar el pronóstico de nuestros pacientes.

