











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
Las displasias esqueléticas o displasias osteocondrales son un grupo heterogéneo de trastornos genéticos que causan anormalidad estructural en el hueso o en el cartílago que llevan a una alteración en el crecimiento de las extremidades o en el tronco, siendo así una de las principales causas de retraso severo del crecimiento 1),(2.
Según la nosología y clasificación actual de los trastornos esqueléticos genéticos (año 2015), se identifican aproximadamente 436 displasias esqueléticas, divididas en 42 grupos en función de características bioquímicas, radiológicas y moleculares 3 . La incidencia de estas entidades es desconocida, pero se estima que aproximadamente se presenta 1 caso por cada 3000 a 5000 recién nacidos vivos 1),(3. La clasificación de acuerdo a la etiología molecular y desórdenes genéticos es la siguiente: 1. Alteración de las proteínas de la matriz del cartílago, 2. Alteración de receptores transmembrana, 3. Alteración del transporte de iones, 4. Alteraciones en factores de transcripción, 5. Defectos en la reabsorción ósea y 6. Defectos desconocidos4.
La patogénesis puede atribuirse a varios factores , entre ellos, a un déficit de componente tisular normal, exceso de alteraciones tisulares secundarias a la mutación, acumulación de metabolitos intermedios e irregularidades en los mecanismos que regulan el desarrollo del tejido osteocartilaginoso.
Dentro del grupo relacionado con alteración de los factores de transcripción se encuentra el Sindrome de Ellis-van Creveld (SEVC), también llamado displasia condroectodermal. Es un trastorno autosómico recesivo poco frecuente, del cual se han reportado aproximadamente 300 casos a nivel mundial. La prevalencia al nacimiento es de 5 a 7 por cada 100.000 nacidos vivos, y se encuentra asociado en 30% de los casos a consanguinidad, siendo más frecuente en las poblaciones como los Amish y en algunas poblaciones árabes 5.
El síndrome es el resultado de mutaciones homocigotas o heterocigotas compuestas en la subunidad 1 del complejo ciliar EvC (EVC) o el genEVC2. A nivel cromosomal, estos genes se ubican en el locus 4p16, el gen EVC y EVC2 tienen 21 y 22 exones codificantes respectivamente. En la mayoría de los casos, las personas afectadas con mutaciones en estos genes tienen el espectro típico de características y no se pueden establecer diferencias fenotípicas para la expresión de los dos genes 5.
Clínicamente, el síndrome se caracteriza por enanismo desproporcionado debido a la condrodisplasia de los huesos largos y un tronco excepcionalmente largo. La severidad del acortamiento de las extremidades aumenta desde la porción proximal a la distal, además se observa polidactilia postaxial bilateral de las manos, ubicado principalmente en el lado cubital y defectos de la cara lateral del extremo proximal de la tibia que conduce a una deformidad genu valgum. En la mayoría de pacientes se observa displasia ectodérmica mostrando uñas y dientes hipoplásicos y anormalmente formados, pelo escaso y delgado. Aproximadamente el 50-60% de los casos presentan un defecto cardíaco estructural siendo los más comunes la aurícula única y defectos del septo ventricular 6.A pesar de la rareza del síndrome, el diagnóstico no debería presentar problemas debido a sus claras manifestaciones clínicas.
El objetivo del presente texto fue presentar un caso del síndrome de Ellis van Creveld descrito de manera integral y llevar a cabo una revisión de la literatura disponible acerca del mismo.
Presentación del caso
Paciente de 12 años, género femenino de procedencia del oriente del departamento del Cauca, remitida por la especialidad de ortopedia por acortamiento de extremidades.
Entre sus antecedentes, durante su gestación, la madre recibió manejo para epilepsia con carbamazepina, ácido valproico, levetiracetam y clobazam. El parto fue por vía vaginal sin complicaciones, si bien no se aportan datos de peso o talla al nacimiento.
En cuanto a los antecedentes personales, la paciente presenta cardiopatía congénita tipo canal auriculoventricular con hipertensión pulmonar, diagnósticada mediante ecocardiograma. Ha sido sometida a cirugías para corrección de polidactilia bilateral en manos y cateterismo cardíaco. Como antecedentes familiares, se tiene que los padres son de la misma región y no presentan apellidos en común.
Al examen físico se encuentra: Peso: 29Kg, Talla: 125cm (T/E<-3 DS) (IMC: +1 a +2 DS), PC: 51 CM, PT: 62cm (%3-25), PA: 60cm, Pabellones: Derecho 5.2x2.4cm(%3-25), Izquierdo 4.5x2.5 cm(<%3), DIC: externa: 110(>97%), interna: 35 mms ( >75%), Distancia intermamilar: 11cm (< %3 ), Filtrum: 11 mm. SS/SI: 1.4 y E-T: -14 . A la inspección, se encuentra nariz y puente nasal anchos, alas nasales pequeñas, frenillo prominente, hiperplasia gingival, hipoplasia de esmalte dental, diastemas, transposición y caries dental, micrognatia. Presencia de tórax estrecho y alargado, escoliosis lumbar leve, escápulas. Soplo holosistólico grado 3/4 con reforzamiento de p2. Abdomen globoso, línea alba hiperpigmentada. Extremidades asimétricas con acortamiento global de predominio acromesomelico. En miembros inferiores valgo severo con asimetría en longitud y deformidad en tijera, rodillas sobrelapadas, braquidactilia con falange proximal normal y falanges medias y distales cortas, clinodactilia, uñas hipoplásicas y onicodistrofia. Pies pequeños, anchos, limitación para la marcha. (Figura 1).
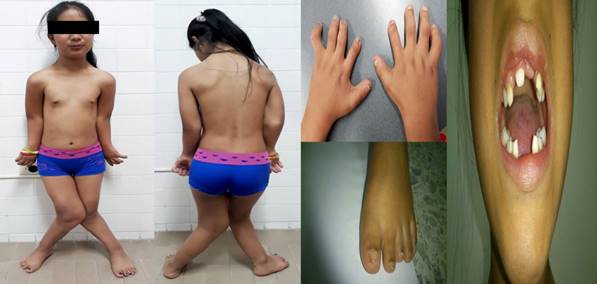
Se llevó a cabo la realización de imágenes diagnósticas, consistentes en radiografías de miembros inferiores y toracolumbar que mostraron escoliosis lumbar de vértice izquierdo en nivel L3-L4 y alteraciones en los núcleos de crecimiento de la tibia de forma bilateral con pérdida de la relación articular femorotibial, con desviación en valgo y lateralización de las rotulas. Densidad ósea y de los tejidos blandos normal (Figura 2).

Con los antecedentes de paciente y con el examen clínico realizado por la especialidad de genética se plantea el diagnóstico del síndrome por lo que se envía a realización de prueba molecular, la cual confirma el diagnóstico (Tabla 1)

Se encuentra actualmente en seguimiento multidisciplinario e interdisciplinario con cardiología por cardiopatía congénita, oftalmología, terapia física y ortopedia quien considera no intervención quirúrgica por alto riesgo relacionado con cardiopatía.
Discusión
El síndrome de Ellis Van Creveld es una patología poco frecuente pero con consecuencias importantes y en algunos casos fatales para los pacientes. El suroccidente de Colombia es una región donde destacan el gran número de grupos étnicos con limitada interacción social, aumentando la probabilidad de endogamia y consanguinidad, lo cual puede conducir a la presentación de patologías poco frecuentes con herencia autosómica recesiva 7),(8.
En nuestro texto presentamos un caso de síndrome de Ellis Van Creveld diagnósticado por la especialidad de genética del hospital de III nivel de la ciudad de Popayán. A nivel mundial se han reportado tan solo 300 casos 5, a nivel nacional y regional no existe epidemiologia de este tipo de síndrome.
La literatura muestra un aumento de la incidencia en grupos étnicos donde la endogamia y cosanguinidad son comunes 8, además, se encontró como antecedente relevante la exposición a sustancias consideradas como tóxicas durante el embarazo (glifosato y anticonvulsivantes). El uso de anticonvulsivantes durante el embarazo se ha asociado a diversas malformaciones congénitas, tales como cardiopatías complejas, reducción de miembros, paladar hendido, microcefalia, polidactilia 9),(10 ; en tanto que la exposición al glifosato durante el embarazo se ha asociado con microcefalia, anencefalia y malformaciones craneales 11 .
Tal y como se observa en nuestra paciente, hay una baja talla patológica de inicio prenatal y detección en la infancia con acortamiento de extremidades que afecta los huesos tubulares y las partes distales resultando en un enanismo acromesomélico aunado a polidactilia bilateral en miembros superiores y en un 10% de los casos asociada a polidactilia en miembros inferiores, hipoplasia y/o ausencia de falanges distales 12), (14.
A nivel torácico, se encuentran costillas cortas y estrechas con pectum excavatum y lordosis lumbar aumentada 15, lo cual se corrobora en nuestra paciente encontrándose tórax largo con diámetro torácico y distancia intermamilar disminuida, además de escoliosis lumbar importante.
A pesar de que a nivel cráneo facial no se han informado malformaciones características del síndrome, existen algunos textos que describen base craneal pequeña, prognatismo, retrognatia y depresión del puente nasal 15,),(16 .Esta última característica se observó en nuestra paciente, sumada al hallazgo de micrognatia y telecanto externo.
Nuestro caso presenta hipoplasia del esmalte dental, dientes pequeños, diastemas, frenillo prominente, sinequia de labio superior, alteraciones descritas de manera amplia en la literatura (17). La alteración oftalmológica principal reportada en la literatura es el estrabismo 18 , el cual no se encontró en nuestra paciente.
La mayoría de las anomalías cardíacas congénitas asociadas al síndrome son defectos del septo auriculoventricular, el cual abarca desde defectos del septo interauricular de tipo ostium primum de tamaño variable hasta la aurícula única. En un 20% de los casos existen otras cardiopatías congénitas como la coartación de aorta, hipoplasia del ventrículo izquierdo o persistencia del ductus arterioso 19 .La distrofia torácica característica de SEVC se asocia con volumen alveolar reducido y taquipnea superficial que acompañado por flujo sanguíneo pulmonar alto secundario a cardiopatías congénitas de base llevan a aumento de resistencias en las vías respiratorias, disminución de la distensibilidad y volumen pulmonar, llevando finalmente a cambios vasculares hipertensivos severos 20. Nuestro caso presenta cardiopatía congénita tipo canal auriculoventricular asociada a hipertensión pulmonar severa. La displasia toráxica se evidencia por las importantes deformidades óseas y físicas en general de nuestra paciente 13),(15),(18
El diagnóstico del síndrome se basa en las características clínicas observadas en los pacientes, el cual se confirma con la prueba molecular basada en homocigosidad de la mutación de genes EVC y EVC2 mediante secuencia directa 21. El diagnóstico aproximadamente se puede hacer desde la semana 18 de desarrollo intrauterino mediante ultrasonografía 22, reportándose retraso de crecimiento, malformaciones esqueléticas y defectos cardiacos 23. Algunos textos hacen referencia a diagnósticos mediante este método en el primer trimestre en donde además de las características mencionadas se observa aumento de grosor de la translucencia nucal 24 .
El diagnóstico genético molecular prenatal se realiza en muestras de vellosidades coriónicas o líquido amniótico 25, en donde se analizan mutaciones de los genes EVC y EVC2, genes adyacentes ubicados en el cromosoma 4p16, dando como resultado alteración y perdida de la función de la proteína 26, análisis fundamental en pacientes quien la clínica genera alta sospecha de la presencia de la patología 24. La literatura reporta estudios de mayor complejidad a los cuales pocas instituciones tienen acceso, tal es el caso de la embriofetoscopia transabdominal con el fin de detectar displasias esqueléticas 24.
El gen EVC codifica una proteína de 992 aminoacidos y el EVC2 una de 1308, son múltiples las mutaciones que se pueden encontrar en estos genes tales como mutaciones puntuales, deleciones, duplicaciones, sitios de corte y empalme exón-intron 27. La proteína codificada denominada Sonic HedgeHog es un morfogeno que regula la organogénesis de verterados, jugando un papel importante en la proliferación de condrocitos de la zona de hipertrofia del cartílago de crecimiento, desarrollo dental, osificación endocondral, desarrollo de extremidades, entre otras 27.
El tratamiento de los pacientes con SEVC requiere atención multidisciplinaria durante el período neonatal y debe incluir manejo con pediatra, ortopedia, cardiología, neumología, odontología y terapeuta ocupacional 28, debido a sus múltiples alteraciones estructurales y funcionales.
En el momento del nacimiento el tratamiento se debe enfocar en el manejo de complicaciones tales como la dificultad respiratoria e insuficiencia cardíaca. Las deformidades de los huesos, en especial el genu valgo necesita un seguimiento ortopédico. Las intervenciones odontológicas, se hacen con el fin de corregir los defectos físicos y funcionales del sistema estomatogmatico 21. La suma de alteraciones a nivel dental y cardiaco han llevado a considerar el uso de antibiótico de manera profiláctica en estos pacientes pero esta indicación aún no se encuentra bien establecida 21.
El pronóstico del SEVC está relacionado con el grado de estrechez torácica y el posible defecto cardíaco. Aproximadamente el 50% de los pacientes con este síndrome fallece en la primera infancia como consecuencia de estas complicaciones. El síndrome no se ha asociado a alteraciones en el desarrollo cognitivo y por tanto los pacientes no presentan alteraciones a este nivel. A nivel familiar, se debe resaltar la importancia del seguimiento por parte de la especialidad de genética con el fin de dar asesoramiento sobre futuros embarazos, dado el riesgo de recurrencia 21),(28.
La prevalencia de síndrome de Ellis Van Creveld es baja pero su diagnóstico no debería causar problemas al personal de salud, ya que sus manifestaciones clínicas son claras y las pruebas diagnósticas disponibles en la actualidad ayudan a confirmarlo. Tal como se observó en el caso presentado en el texto, la atención multi e interdisciplinaria es crucial en la mejoría de la calidad de vida del paciente y el diagnóstico temprano, inclusive en etapa prenatal, contribuye a disminuir la incidencia de complicaciones y al inicio temprano del manejo de alteraciones que aporten al bienestar del paciente y mejoran la morbimortalidad de los mismos. Factores de riesgo como la consanguinidad entre padres aumentan la probabilidad de la ocurrencia de este y otros síndromes raros, por lo tanto se deben utilizar estrategias de promoción y prevención en salud para disminuir la incidencia del mismo

