











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
In recent years, metal coordination complexes have been found to have various physicochemical properties which have been exploited in several fields such as catalysis, the synthesis of novel materials and medicinal chemistry [1-4]. Within the medicinal chemistry, many investigations have been conducted to find new antimicrobial agents; in these studies, several metal complexes have been identified as compounds of interest; metal complexes whose biological activities occur via different mechanisms [4]. Previous studies have been carried out with the aim of discovering alternative treatments for several diseases and fighting antimicrobial resistance, which is a major issue within the field of infectious diseases. This phenomenon is currently increasing and it is now considered a global public health problem [5].
Thiosemicarbazones are Schiff bases, and they have shown potential biological activity against a large range of microorganisms that cause infectious disease, especially bacteria [6]. These types of molecules have also exhibited potent activity against some viruses and multiple types of neoplastic cells [7-9]. The use of Schiff bases for the synthesis of new coordination compounds for biological applications has been widely studied because of their electronic properties, allowing the presence of different coordination sites (electron donor groups, N and S) to which metal ions can bind. Metals bound to these groups can lead to a synergistic effect that improves the biological activity (Fig- 1) [10,11].
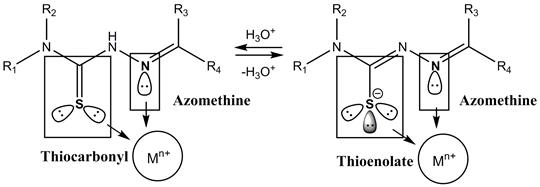
Figure 1 General structure of the thiosemicarbazones and coordination sites with the metal center (Mn+).
Lanthanide complexes have also gained substantial interest for therapeutic applications. The unusual electronic configuration of the lanthanides gives them several features such as fluorescence and redox behavior that can be of high importance for some medicinal treatments including contrast, antimicrobial and chemotherapeutic agents [1,2,12].
Additionally, the studies on the interactions between metal complexes and DNA strands have offered some ideas regarding the possible mechanisms of action; numerous compounds that are cytotoxic to tumoral cells can bind to DNA strands. Thereby, the evaluation of the interactions that these types of molecules may have with the DNA provides an initial understanding of their biological activity [13-17].
Thus, considering the potential therapeutic uses of thiosemicarbazones and lanthanide complexes and in order to contribute to the discovery of new molecules able to fight antimicrobial resistance, the aim of this research is to describe the synthesis and characterization of La(III), Nd(III) and Eu(HI) complexes of N(4)-(4-R-phenyl) thiosemicarbazones derived from the 2-carboxybenzaldehyde. Additionally, to evaluate the interactions between DNA and the complexes by different techniques and to discuss the antibacterial assays.
Material and methods
All reagents for the synthesis were purchased from Sigma Aldrich, Alfa Aesar and Merck and were used without further purification. All solvents used were of analytical grade. The infrared spectra were obtained between 4000 cm-1 and 600 cm-1 on a Shimadzu Affinity 1 (FT-IR) device with an ATR accessory. For the inorganic region (between 600 cm-1 and 250 cm-1) a NICOLET 6700 (FT-IR) Thermo Scientific spectrophotometer was used. IR spectra were acquired using from KBr pellets. Elemental analyses were carried out using a Thermo Scientific EA 112 series CHN Analyzer; and the lanthanide percentage was determined by complexometric titration with EDTA. Melting points were determined using an automatic OptiMelt MPA100 device. Conductivity measurements of the lanthanide complexes were performed in an Orion™ 131S. 1H-NMR and 13C-NMR spectra were recorded in DMSO-d6 at 25 °C using a Bruker Ultrashield Avance II400 spectrophotometer operating at 400 MHz, and chemical shifts (δ) are expressed in units of ppm with respect to (tetramethylsilane) TMS. The overall assignment of the NMR signals for the new compounds was supported by analyses of their 2D spectra (HMBC and HSQC). Thermal analyses were performed in a TGA 550 TA Instruments device under a nitrogen atmosphere. Mass spectra were taken with a Shimadzu-GCMS-QP2010 device with electronic impact ionization at 40 eV.
Synthesis of ligands
The syntheses of 2-carboxybenzaldehyde N(4)-phenylthiosemicarbazone (L1) and 2-carboxybenzaldehyde N(4)-(4-chlorophenyl) thiosemicarbazone (L2) were carried out as shown in Fig. 2. The syntheses were conducted following the standard methodologies that were previously reported [6,18-22].
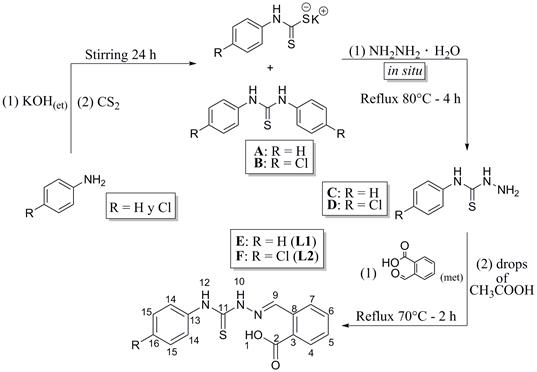
Synthesis of 2-carboxybenzaldehyde N(4)-phenylthiosemicarbazone (L1)
An ethanolic solution (3 mL) of aniline (0.55 mol-L-1, 1.64 mmol) and potassium hydroxide (0.55 mol-L-1, 1.64 mmol) was slowly added to 2 mL of carbon disulfide (33.1 mmol, excess) in an ice bath (4 °C), and the formation of two phases was observed immediately, and the mixture was stirred constantly for 24 hours at room temperature. Once the reaction was complete and a white solid in a yellow solution had been obtained, 80 μ of hydrazine monohydrate (1.65 mmol) was added, and the mixture was refluxed at 80 °C for 4 hours. Then, the solution was concentrated to 3 mL and cooled to 4 °C in an ice bath. Once cool, 5 mL of a cold, 1:1 mixture of hexane and dichloromethane was added to precipitate the N(4)-phenylthiosemicarbazide, and the solid was isolated by filtration and washed with 8 mL of cold hexane.
A methanolic solution (2.5 mL) of 2-carboxybenzaldehyde (0.48 mol-L-1, 1.20 mmol) and three drops of glacial acetic acid were added to a methanolic solution (2.5 mL) of N(4)-phenylthiosemicarbazide (0.48 mol-L-1, 1.20 mmol), and the system was refluxed at 70 °C for 2 hours. Once the reaction was complete, a white solid was observed (L1); the excess solvent was evaporated under reduced pressure, and the product was washed with 8 mL of cold hexane 3 times. White powder (yield: 341.3 mg, 95 %). m.p.: 203 - 204 °C.
C15H13N3O2S, Elemental Analysis; C, 59.92 (calc. 60.19); H, 4.32 (4.38); N, 13.97 (14.04); S, 9.57 (10.71) %. IR (ATRcm-1) v: 3331 m, 3181 m, 3014 m, 2831 w, 2577 w, 2456 w, 2360 w, 2341 w, 1666 s, 1601 w, 1589 w, 1545 m, 1515 m, 1481 m, 1389 m, 1240 s, 1202 s, 1068 s, 943 m, 820 m, 793 m, 745 s, 694 s. 1H-NMR (DMSO-d6, 400 MHz): δ H 7.20 (t, J = 7.4 Hz, 1H, H16), 7.37 (t, J = 7.9 Hz, 2H, H15), 7.51 (td, J = 7.5 Hz, 1.2 Hz, 1H, H5), 7.59 (d, J = 8.2 Hz, 2H, H14), 7.61 (td, overlapped with H14, 1H, H6), 7.86 (d, J = 7.7 Hz, IH, H7), 8.38 (d, J = 7.9 Hz, 1H, H4), 8.90 (s, 1H, H9), 10.08 (s, 1H, H12), II. 98 (s, 1H, H10), 13.32 (s, 1H, H1). 13C-NMR (DMSO-d6, 100 MHz): δ C 125.3 (C16), 125.7 (C14), 127.5 (C4), 128.1 (C15), 129.6 (C5), 130.0 (C7), 131.0 (C3), 131.6 (C6), 134.0 (C8), 139.1 (C13), 142.1 (C9), 168.3 (C2), 176.2 (C11). MS (EI, 40 eV): m/z 299 [M+].
Synthesis of 2-carboxybenzaldehyde N(4)-(4-chlorophenyl) thiosemicarbazone (L2)
An ethanolic solution (3 mL) of aniline (0.67 mol-L-1, 2.00 mmol) and potassium hydroxide (0.67 mol-L-1, 2.00 mmol) was slowly added to 2 mL of carbon disulfide (33.1 mmol, excess) in an ice bath (4 °C), the formation of two phases was immediately observed, and the mixture was stirred constantly for 24 hours at room temperature. Once the reaction was completed and a white solid in a yellow solution had been obtained and 97 μL (2.00 mmol) of hydrazine monohydrate had been added, the mixture was refluxed at 80 °C for 4 hours. Then, the solution was concentrated to 3 mL and cooled to 4 °C in an ice bath, 5 mL of a cold, 1:1 mixture of hexane and dichloromethane was added to precipitate the N(4)-(4-chlorophenyl)thiosemicarbazide. The solid was isolated by filtration and washed with 8 mL of cold hexane.
A methanolic solution (2.5 mL) of 2-carboxybenzaldehyde (0.556 mol-L-1, 1.39 mmol) and four drops of glacial acetic acid were added to a methanolic solution (2.5 mL) of N(4)-(4-chlorophenyl)thiosemicarbazide (0.556 mol-L-1, 1.39 mmol), and the system was refluxed at 70 °C for 2 hours. Once the reaction was complete, a white solid was observed (L2), the solvent excess was evaporated under reduced pressure and the product was washed with 8 mL of cold hexane 3 times. Pale yellow powder (yield: 634.2 mg, 95 %). m.p.: 215-216 °C. C15H12N3O2SCl, Elemental Analysis; C, 54.12 (calc. 53.98); H, 3.60 (3.62); N, 12.42 (12.59); S, 9.20 (9.60)%. IR (ATR cm-1) v: 3332 w, 3176 m, 3015 m, 2588 w, 2448 w, 1670 s, 1604 w, 1588 w, 1548 s, 1512 m, 1486 m, 1383 m, 1266 m, 1226 s, 1194 s, 1069 s, 1009 m, 950 w, 821 s, 790 m, 757 m, 670 m, 632 m. 1H-NMR (DMSO-d6, 400 MHz): δ H 7.42 (d, J = 8.7 Hz, 2H, H14), 7.51 (td, J = 7.5 Hz, 0.9 Hz, 1H, H5), 7.60 (t, J = 7.4 Hz, 1H, H6), 7.63 (d, J = 8.7 Hz, 2H, H15), 7.87 (dd, J = 8.1 Hz, 1.2 Hz, 1H, H7), 8.38 (d, J = 7.7 Hz, 1H, H4), 8.92 (s, 1H, H9), 10.13 (s, 1H, H12), 12.06 (s, 1H, H10), 13.33 (s, 1H, H1). 13C-NMR (DMSO-d6, 100 MHz): δC 127.3 (C15), 127.5 (C4), 127.9 (C14), 129.3 (C16), 129.6 (C5), 130.0 (C7), 131.0 (C3), 131.6 (C6), 134.0 (C8), 138.0 (C13), 142.4 (C9), 168.2 (C2), 176.2 (C11). MS (EI, 40 eV): m/z 333 [M+].
Synthesis of the lanthanide complexes
The syntheses of the lanthanide complexes were carried out with carboxylic acids in aqueous solutions according to methodologies reported in previous investigations (Fig. 3.) [1,2,19]. The numeration of the protons in the complexes is the same as the one employed for the ligands.

Synthesis of [La(L1)2 (Cl)].2H2 O (1)
Distilled water (14 mL) was added to 100 mg (0.330 mmol) of 2-carboxybenzaldehyde N(4)-phenylthiosemicarbazone, and the pH was adjusted to between 7 and 7.5 until all the solid was dissolved. Then, an aqueous solution (5 mL) of LaCl3-7H2O (0.033 mol-L-1, 0.165 mmol) was added dropwise to the ligand solution. A white solid immediately appeared, and the mixture was stirred for 4 hours at 50 ° C. Finally, the white solid was isolated by filtration and washed with 10 mL of distilled water and 10 mL of cold hexane. The compound was then placed in a desiccator for 2 days. White powder (yield: 242.6 mg, 90 %). m.p.: 208 - 209 °C. C30H28N6LaO6S2Cl, Elemental Analysis; C, 44.06 (calc. 44.65); H, 3.69 (3.50); N, 11.54 (10.41); S, 7.71 (7.94); La, 17.27 (17.21) %. IR (KBr pellet cm-1) v: 3327 w, 3195 w, 1594 w, 1572 w, 1548 s, 1532 s, 1498 m, 1473 w, 1447 m, 1394 s, 1273 m,1195 m, 1195 m, 1068 w, 938 w, 755 m, 744 m, 691 m, 463 w. 1H-NMR (DMSO-d6, 400 MHz): δH 7.18 (t, J = 7.3 Hz, 2H, H16), 7.35 (t, J = 7.7 Hz, 4H, H15), 7.40 - 7.42 (m, 4H, H5 and H6), 7.59 (d, J = 7.5 Hz, 4H, H14), 7.93 (d, J = 7.7 Hz, 2H, H7), 8.17 (d, J = 8.4 Hz, 2H, H4), 9.19 (s, 2H, H9), 10.03 (s, 2H, H12), 11.71 (s, 2H, H10). 13C-NMR (DMSO-d6, 100 MHz): δC 125.2, 126.6, 126.8, 128.1, 128.6, 128.8, 129.2, 129.6, 130.2, 139.2, 145.1, 176.0. A (DMSO, 28 °C) (Ω-1-cm2-mol-1): 17.6.
Synthesis of [La(L2)2(Cl)]·4H2O (2)
Distilled water (14 mL) was added to 200 mg (0.600 mmol) of 2-carboxybenzaldehyde N(4)-(4-chlorophenyl)thiosemicarbazone, and the pH was adjusted to between 7 and 7.5 until all the solid dissolved. Then, an aqueous solution (5 mL) of LaCl3-7H2O (0.060 mol-L-1, 0.300 mmol) was added dropwise to the ligand solution. A white solid immediately appeared, and the mixture was stirred for 4 hours at 50 °C. Finally, the white solid was isolated by filtration and washed with 10 mL of distilled water and 10 mL of cold hexane. The compound was then placed in a desiccator for 2 days. White powder (yield: 459.0 mg, 84%). m.p.: 205 - 206 °C. C30H30N6LaO8S2Cl3, Elemental Analysis; C, 39.97 (calc. 39.51); H, 3.80 (3.32); N, 9.83 (9.22); S, 6.75 (7.03); La, 15.19 (15.23) %. IR (KBr pellet cm-1) v: 3317 m, 1584 w, 1571 w, 1517 s, 1451 m, 1412 m, 1395 s, 1287 m, 1263 m, 1187 m, 1069 m, 1010 m, 938 m, 848 w, 829 m, 808 w, 768 m, 755 m, 680 m, 451 w. 1H-NMR (DMSO-d6, 400 MHz): 8 H 7.35-7.39 (m, 8H, H5, H6 and H14), 7.52 (d, J = 8.6 Hz, 4H, H15), 7.72-7.74 (m, 2H, H7), 8.09- 8.11 (m, 2H, H4), 8.95 (s, 2H, H9), 10.00 (s, 2H, H12), 11.55 (s, 2H, H10). 13C-NMR (DMSO-d6,100 MHz): δC 128.3, 128.5, 129.6, 130.4, 130.5, 131.2, 132.9, 133.6, 134.5, 138.5, 145.7, 167.4, 176.7. A (DMSO, 28 °C) (Ω-1-cm2-mol-1): 17.0.
Synthesis of [Nd(L1)2(Cl)] (3)
The synthesis of 3 was conducted in a manner identical to the synthesis of 1. Pale violet powder (yield: 207.5 mg, 80%). m.p.: 207 - 208 °C. C30H22N6NdO4S2Cl, Elemental Analysis; C, 48.70 (calc. 46.41); H, 3.67 (3.12); N, 11.26 (10.82); S, 7.41 (8.26); Nd, 18.88 (18.58) %. IR (KBr pellet cm-1) v: 3299 w, 1595 w, 1561 w, 1545 m, 1496 m, 1471 w, 1445 m, 1392 s, 1269 w, 1187 m, 1070 w, 940 w, 754 m, 744 s, 690 m, 453 w. 1H-NMR (DMSO-d6, 400 MHz): δ H 7.2 (t, J = 7.6 Hz, 2H, H16), 7.37 (t, J = 7.5 Hz, 4H, H15), 7.62 (d, J = 7.4 Hz, 4H, H14), 7.80 (br, 2H, H5), 7.94 (br, 2H, H6), 8.62 (d, J = 6.4 Hz, 2H, H7), 9.59 (br, 2H, H4), 10.16 (s, 2H, H12), 10.29 (br, 2H, H9), 11.65 (s, 2H, H10). 13C-NMR (DMSO-d6, 100 MHz): δC 124.9,125.4, 127.8, 128.0, 129.8, 130.1, 131.0, 27 145.3. A (DMSO, 28 °C) (Ω-1-cm2-mol-1): 14.4.
Synthesis of [Nd(L2)2(Cl)] (4)
The synthesis of 4 was carried out in a manner identical to the synthesis of 2. Pale violet powder (yield: 436.1 mg, 86%). m.p.: 203 - 204 °C. C30H22N6NdO4S2Cl3, Elemental Analysis; C, 42.57 (calc. 42.63); H, 3.03 (2.62); N, 10.28 (9.94); S, 7.32 (7.59); Nd, 17.19 (17.06) %. IR (KBr pellet cm-1) v: 3316 m, 1583 m, 1555 w, 1520 s, 1479 m, 1449 m, 1415 s, 1395 s, 1341 w, 1263 m, 1188 m, 1088 m, 1070 m, 848 m, 828 m, 808 m, 768 m, 756 m, 684 w, 460 m. 1H-NMR (DMSO-d6, 400 MHz): δ H 7.12 - 8.21 (br, H chlorophenyl moiety), 8.23 - 8.68 (br, 4H, H5), 9.17 - 9.74 (br, 4H, H6), 10.01 - 10.46 (br, 6H, H9, H12 and H 2-carboxybenzil moiety), 11.71 (s, 2H, H10). 13C-NMR (DMSO-d6,100 MHz): δc 127.0,127.6, 128.1, 129.7, 130.0, 131.0. A (DMSO, 28 °C) (Ω -1-cm2-mol-1): 12.2.
Synthesis of [Eu(L1)2(Cl)] (5)
The synthesis of 5 was performed in a manner identical to the synthesis of 1. Pale yellow powder (yield: 238.4 mg, 91%). m.p.: 206 - 207 °C. C30H22N6EuO4S2Cl, Elemental Analysis; C, 46.78 (calc. 45.95); H, 3.59 (3.09); N, 11.11 (10.72); S, 7.27 (8.18); Eu, 18.59 (19.00) %. IR (KBr pellet cm-1) v: 3314 w, 1595 m, 1575 w, 1524 s, 1496 m, 1475 m, 1445 m, 1393 s, 1327 w, 1268 m, 1187 m, 1069 w, 941 w, 757 m, 745 s, 691 m, 640 w, 407 m. 1H-NMR (DMSO-d6, 400 MHz): δ H 6.20 (s, 2H, H5), 6.80 (s, 2H, H6), 7.07 (s, 2H, H7), 7.16 (t, J =7.2 Hz, 2H, H16), 7.32 (t, J = 7.5 Hz, 4H, H15), 7.45 (br, 2H, H9), 7.54 (d, J = 7.5 Hz, 4H, H14), 7.64 (d, J = 5.9 Hz, 2H, H4), 9.86 (s, 2H, H12), 11.72 (s, 2H, H10). 13C-NMR (DMSO-d6,100 MHz): δc 125.1, 125.6, 128.0, 128.3, 129.2, 132.9, 139.1, 144.2, 175.9. A (DMSO, 28 °C) (Ω -1-cm2-mol-1): 13.9.
Synthesis of [Eu(L2)2(Cl)]·H2O (6)
The synthesis of 6 was carried out in a manner identical to the synthesis of 2. Pale yellow powder (yield: 417.5 mg, 80%). m.p.: 213 - 214 °C. C30H24N6EuO5S2Cl3, Elemental Analysis; C, 41.57 (calc. 41.37); H, 3.07 (2.78); N, 9.93 (9.65); S, 7.16 (7.36); Eu, 18.24 (17.85) %. IR (KBr pellet cm-1) v: 3320 m, 1584 w, 1563 w, 1522 s, 1477 m, 1449 w, 1395 s, 1324 w, 1287 m, 1262 m, 1186 s, 1089 m, 1070 m, 1011 m, 940 m, 849 w, 828 m, 808 w, 768 m, 756 m, 455 m. 1H-NMR (DMSO-d6, 400 MHz): 8 H 6.18 (br, 2H), 6.82 - 7.56 (m, 18H), 9.92 (s, 2H, H12), 11.74 (s, 2H, H10). 13C-NMR (DMSO-d6,100 MHz): δc 127.2, 127.9, 136.0. A (DMSO, 28 °C) (Ω -1-cm2-mol-1): 11.4.
DNA interaction assays
The DNA interaction studies with the obtained compounds were accomplished by electronic absorption experiments, oxidative cleavage monitored by agarose gel electrophoresis and viscosity measurements. Highly polymerized and lyophilized calf thymus DNA (CT-DNA) from Sigma Aldrich was employed, and the pBR322 vector purchased from Promega was also used. All the DNA solutions had an A260/A280 ratio between 1.8 and 1.9, indicating that the DNA was free of RNA and proteins. DNA resuspensions were carried out using a solution of Tris (10 mmol-L-1) and EDTA (1 mmol-L-1) in deionized water with the pH adjusted to approximately 7.5. The DNA solutions were stored at -5 °C.
Electronic absorption monitoring assays (UV-Vis)
The electronic absorption spectra were recorded in an UV-visible Jasco V-630BIO spectrophotometer equipped with a PAC-743 accessory for temperature control. CT-DNA titrations were carried out to a constant concentration of compound at 25 °C. After each addition of titrant, the electronic absorption spectra of the evaluated compound was taken between 230 nm and 500 nm. A CT-DNA solution (4022 μmol-L-1) in nucleotides was prepared using ε 260 = 6600 L·cm-1-mol-1- as the molar extinction coefficient and measuring the absorbance at 260 nm; the stock solution was stored at -5 °C.
Oxidative cleavage assays monitored by agarose gel electrophoresis
The oxidative cleavage catalyzed by the lanthanide complexes was studied by incubating different solutions (24 μL each) that contained constant concentrations of pBR322 and H2O2 with variable concentrations of the complexes in DMSO. The samples were incubated under physiological conditions (37 °C and pH 7.0) in an Eppendorf Thermomixer® C for 90 minutes. Once incubated, the samples were loaded onto a 1 % agarose gel with TAE 1X buffer, and HyperLadder™ 1 kb from Bioline used as the molecular weight marker. An OwlTM EasyCast™ B1 Mini Gel electrophoresis system was used for the electrophoresis runs, and GelGreen™ was used as the dye to visualize the gel in an UltraBright Led 470 nm MaestroGen transilluminator.
Viscosity measurements
The relative viscosities of the five different DNA solutions were determined. The solutions contained a constant concentration of CT-DNA and increasing amounts of the test compounds. A stock solution of CT-DNA in nucleotides (402 μmol·L-1) and a Semi-Micro Cannon-Ubbelohde viscometer 75 were used for determining the viscosity; the temperature of the solutions was controlled by using a thermal bath at 22 °C. The efflux times were recorded by processing the videos photograms in Camtasia Studio® 8 software.
Antibacterial assays
The antibacterial activities of all the compounds were evaluated by determining their minimal inhibitory concentrations (MICs); and standard microdilution procedures for antimicrobial susceptibility were followed [23,24]. All compounds were tested against five different ATCC bacterial strains: ATCC 25923 (Staphylococcus Aureus), ATCC 25922 (Escherichia Coll), ATCC 14028 (Salmonella Typhimurium), ATCC 2146 and ATCC 1705 (Klebsiella Pneumoniae); the evaluated concentration range was from 2000 [ug-mL to 4 μg-mL. The negative control consisted of Muller-Hinton broth (MHB) in the absence of bacteria, the positive control consisted of MHB with only bacteria, and no commercial bactericide was employed as reference. An additional study was carried out to evaluate the possible effect of the solvent on the biological screening, and it was observed that DMSO did not show any activity against any of the tested strains.
Results and discussion
Synthesis and characterization of ligands
Thiosemicarbazones were obtained through the nucleophilic addition of the corresponding para-substituted anilines to the carbon disulphide; in this step, the thiocarbamate potassium salt was synthesized in situ. The white solids that precipitated at this stage were identified as 1.3- diphenylthiourea (see Fig. 2a) (MS (EI, 40 eV): [M+] m/z 228) and 1.3- bis (4-chlorophenyl)thiourea (see Fig. 2b) (MS (EI, 40 eV): [M+] m/z 297), which were also useful for the synthesis of thiosemicarbazides (see Fig. 2c and Fig. 2d) by the substitution with monohydrated hydrazine. The identification of N(4)-phenylthiosemicarbazide (m.p.: 140 -141 °C) and N(4)-(4- chlorophenyl) thiosemicarbazide (m.p.: 165 -167 °C) was conducted by comparing their melting points and IR absorption bands with those reported in previous investigations [18,20]. After the Schiff base condensation with 2-carboxybenzaldehyde, novel ligands L1 and L2 were obtained as solid powders. They were soluble in DMSO, acetone, DMF and ethyl acetate, and they were slightly soluble in water, ethanol and methanol.
Synthesis and characterization of lanthanide complexes
All lanthanide complexes (1-6) were isolated as solid powders. They were soluble in DMSO, acetone and DMF, and they were slightly soluble in water, ethanol, methanol and ethyl acetate. The molar conductivities of the complexes in DMSO at 28 °C were found to be between 10 Ω-1-cm2-mol-1 and 18 Ω-1-cm2-mol-1, which confirmed the absence of chloride ions (Cl-), the non-electrolytic nature of the complexes and the possible coordination of the chloride atom to the metal center [18,25]. Furthermore, the elemental analyses of the complexes showed a 1:2 (metal to ligand) stoichiometry.
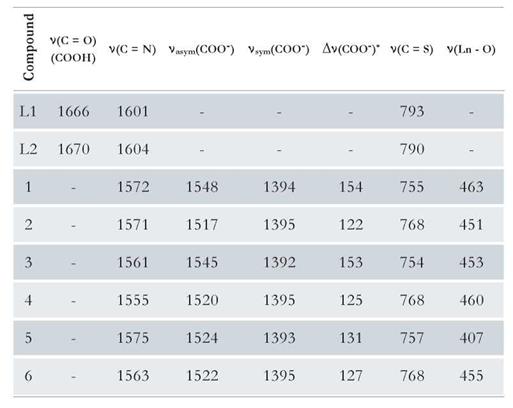
Moreover, the infrared spectroscopic characterization provided important information about the coordination sites in the lanthanide complexes. The shifts to lower energy occurred in the vibrational bands corresponding to the thiocarbonyl (C=S) and azomethine (C=N) groups, which confirmed the participation of these as coordination sites. In addition, the absence of the bands typical of a carboxyl (COOH) group also indicated the coordination to the metal ion. The differences between the symmetric and asymmetric vibrational modes of the carboxylate group in the complexes were between 120 cm-1 and 160 cm-1; this could indicate that the functional group is binding in a bidentate manner. Table 1 shows the most characteristic frequency assignments for each compound [1,17,18,26].
Table 1 Assignments of the characteristic IR absorption bands for the ligands and lanthanide complexes (cm-1).
All proposed molecular formulas for the complexes are consistent with the percentages found in the elemental analyses, and the presented water molecules are placed in the external coordination sphere as waters of hydration according to the thermal analyses. Fig. 4 and Fig. 5 show the thermal behaviors of complexes 1 and 6 as representative complexes.
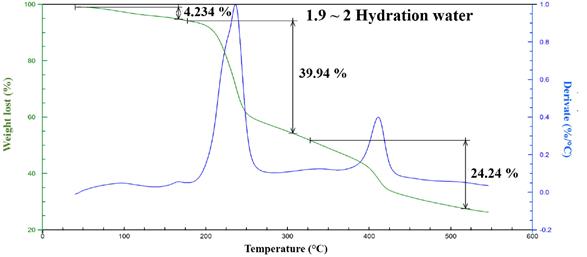

The presence of water molecules in the external coordination sphere was confirmed only for complexes 1, 2 and 6 . All the compounds were highly stable up to 200 ° C at which point they thermally decomposed. The first step in this process seems to be the loss of one of the ligands at approximately 230 °C, which coincides with the mass loss observed at that stage. At approximately 300 ° C, another thermal degradation event is observed, and in this last stage, the loss of the second ligand is likely and leads to the formation of a carbonate derivate [1,2].
The complexes (except those of La(III)) are paramagnetic. 1H-NMR and 13C-NMR analyses were performed for La(III), Nd(III) and Eu(III) complexes, and the results confirmed that thiocarbonyl, azomethine and carboxylate participate in coordination to the lanthanide. The absence of the H1 signal in the spectra is an evidence that the proton had been replaced by the metal ion.
The signals of H9, H10 and H12 are shifted due to the presence of the metal; for complexes 1 and 2, the signal of H9 is shifted downfield due to the inductive effects after the coordination of the azomethine group (C=N). The signals of H10 and H12 are shifted upheld due to the decrease in the inductive effects form the thiocarbonyl group (C=S) once it participates in metal coordination. Similarly, the signals of H10 and H12 for complexes 5 and 6 are shifted upheld relative to the corresponding signals of the ligands L1 and L2; however, the H9 signal is also shifted upheld, indicating that the Eu(III) complexes may be in a different spatial arrangement with the H9 protons possibly facing the metal center (electron source); although, the participation of the azomethine (C=N) in the coordination was confirmed by the IR spectra. Additional shifts in the proton signals assigned to aromatic systems were found for all complexes due to the different chemical environments of the complexes compared to those in the ligands. The broadening of the NMR signals of complexes 4 and 6 may be a consequence of the paramagnetic character of the lanthanide ions, Nd (III) and Eu (III), which have very short relaxation times due to the small difference between the ground state and the excited state energies. In addition, for the complexes with L2, a slight decrease in solubility was observed; thus, the signal can be a mixture of absorption and dispersion. Fig. 6 shows the changes in the chemical shifts for complexes 1-6 with respect to their precursors.
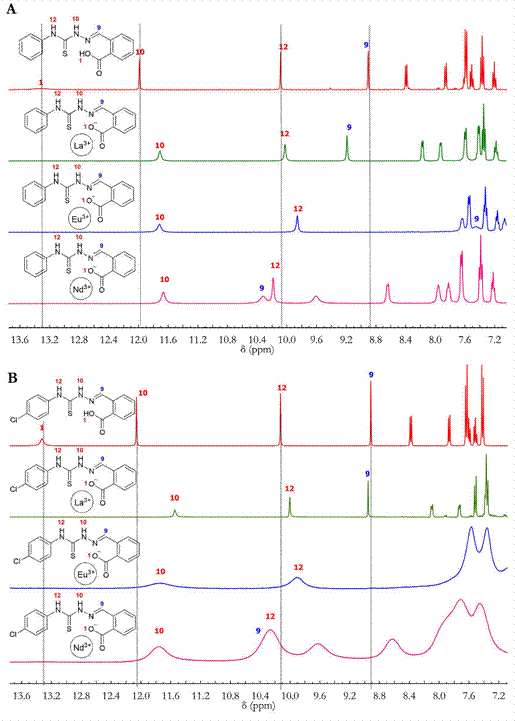
Figure 6 A) 1H-NMR spectra superposition of L1 (red), 1 (green), 3 (purple) and 5 (blue); B) 1H-NMR spectra superposition of L2 (red), 2 (green), 4 (purple) and 6 (blue).
The signal assignments were supported by the 2D experiments (HSQC and HMBC). The 13C-NMR spectra also showed shifts in signals of C2, C11 and C9, which supports the participation of the carboxylate (COO-), thiocarbonyl (C=S) and azomethine (C=N) groups in coordination.
DNA interaction assays
The mode of interaction between the DNA strands and the new molecules with biological applications provides an initial indication of its possible biological activity and mechanism of action. On one hand, the binding ability of the studied molecule is a clear signal of its competence for biological applications [17], because the presence of this type of interaction leads to a cytotoxic effect that causes cell apoptosis [14]; however, the absence of this ability does not mean that the evaluated molecule must be discarded as it could exhibit its activity through a different mechanism such as enzymes inhibition or blocking the membrane receptor [27]. Thus, the assays for determining the mode of interaction between the DNA and ligands L1 and L2 and complexes 1-6 were carried out to determine their binding modes and catalytic abilities in the oxidative cleavage of the strands.
Electronic absorption monitoring assays (UV-Vis)
Figure 7 shows the effect of the addition of CT-DNA between 5 μmol-L-1 to 40 mol-L-1 in nucleotides to a solution of the compounds at 20 μmol-L-1. The solutions of the complexes contained approximately 10 % DMSO to improve the solubilities of the compounds in the resuspension buffer.
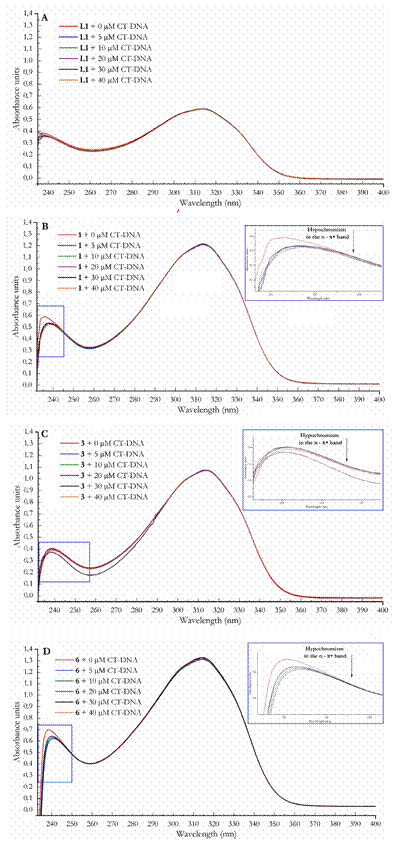
Figure 7 Electronic absorption spectra for (A) L1; (B) L2; (C) 1; (D) 2; (E) 3; (F) 4; (G) 5 and (H) 6 (red) at a constant concentration of 20 μmol-L-1 titrated with CT-DNA to concentrations of 5 μmol-L-1 (blue), 10 μmol-L-1 (green), 20 μmol-L-1 (violet), 30 μmol.L-1 (black) and 40 μmol-L-1 (orange) in nucleotides.
Monitoring the electronic absorption spectra of the studied compound when adding DNA was one of the techniques used to observe the interactions between these two species. The presence of hypochromism and bathochromism is an indicator of nitrogen base intercalation; this is due to the intercalation of the compound between the nitrogen base pairs which hide its chromophores; thus, changing the molecular orbitals, causing minor exposure to radiation and reducing the molar extinction coefficient [15]. Otherwise, a hyperchromic effect is a sign of a change in the tertiary structure of the biopolymer promoted by the compound and could be evidence of DNA scission; this effect is caused by the major exposure of the nitrogen base pairs when unrolling or splitting [28-30].
No net effect was observed for the bands at approximately 315 nm (π → π*) in all the compounds, which suggests that the aromatic groups are not intercalating between the base pairs. However, for complexes 1, 3 and 6, a slight redshift around the 240 nm (n→ π*) is observed. This shift could indicate binding to the DNA groove [17,31].
This redshift can be explained by considering that electron donor groups are responsible for the n→ π* transitions, and these groups are involved in metal coordination and are also the one nearest to the metal, which more closely interacts with the phosphate chain of the biopolymer grooves [28].
Viscosity measurements
The viscosity of a DNA solution at a certain temperature can change if its structure is altered [32]. To observe the compounds’ effect on DNA viscosity, five CT-DNA solutions at 80 μmol-L-1 in nucleotides (with different compound quantities) were evaluated. Here, as in the previous analysis, the solutions contained some amount of DMSO below 10 % to solubilize the compounds in the resuspension buffer.
Figure 8 shows the changes in the viscosities of the CT-DNA solutions in the presence of the evaluated compounds. The viscosities were calculated by finding the ratio between the kinetic viscosities of the mixtures and that of free DNA using the viscometer constant and efflux times.
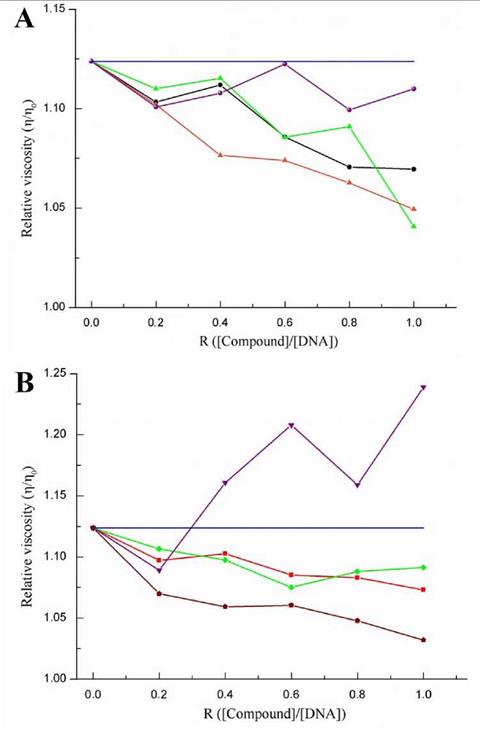
Figure 8 Effect of increasing amounts of compound (R = [compound]/[DNA]) between 0.2 and 1) on the relative viscosities of the CT-DNA solutions at 80 μmol-L-1 (blue line). (A) L1 (black), 1 (green), 3 (violet) and 5 (orange); (B) L2 (red), 2 (green), 4 (violet) and 6 (brown).
The general trend for compounds L1, L2, and complexes 1, 2, 5 and 6 is that they reduce the relative viscosity of CT-DNA; however, the Nd(III) complexes did not seem to follow any certain pattern. The reduction in the CT-DNA viscosity can mainly be attributed to two three-dimensional alterations in the DNA structure. The first one is the reduction in the size of some linear sections of the DNA caused by a possible rolling; this results in a lower probability of intermolecular crossing, thus, reducing the viscosity. The second cause is DNA cleavage in which the solution with free nucleotides possess a lower viscosity than that with the entire biopolymer [30]. For the case of the evaluated compounds, it is possible that the DNA interactions are occurring through groove binding, which induces partial rolling and no DNA cleavage. This was confirmed because if there was DNA scission occurring, a strong hyperchromism would be observed in all the electronic absorption spectra. However, the changes in the DNA viscosity caused by all the compounds are small in comparison with other types of molecules evaluated in other investigations [17,29-33]. The strongest interactions were observed for Eu(III) complexes 5 and 6, suggesting that reducing the ionic radio affects the strength of the interactions. This change is because the Pearson acidity increased, enhancing the affinity for the phosphate ions in the DNA backbone.
Oxidative cleavage assays monitored by agarose gel electrophoresis
The catalytic effects of L1, L2, and complexes 1, 2, 5 and 6 in the cleavage of DNA strands, which is initiated by reactive oxygen species (ROS), were tested by agarose gel electrophoresis. Fig. 9 shows the preference of one of the three pBR322 vector conformations when incubating in several solutions containing plasmid, hydrogen peroxide (ROS generator) and different amounts of the test compounds.
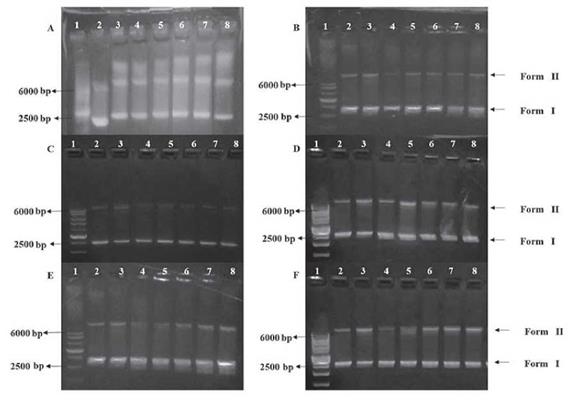
Figure 9 Agarose gel electrophoresis pattern showing the oxidative cleavage of the plasmid pBR322 (5 ng-mL-1) with H2O2 (500 μmol-L-1) and various concentrations of the test compounds. Lane 1: HyperLadder 1 kb (10 μmL); lane 2: only DNA; lane 3: DNA + H2O2; lane 4: DNA + compound (60 μmol-L-1); lane 5: DNA + H2O2 + compound (20 μmol-L-1); lane 6: DNA + H2O2 + compound (50 μmol-L-1); lane 7: DNA + H2O2 + compound (70 μmol-L-1); lane 8: DNA + H2O2 + compound (100 μmol-L-1). (A) L1; (B) L2; (C) 3; (D) 4; (E) 5 and (F)6.
Plasmids are circular DNA that can adopt several forms or conformations. Form I corresponds to the rolled or bent plasmid, which possesses great mobility in agarose gel; form II is the relaxed circular conformation, which has lower mobility than form I, because the circular form gets stuck more frequently in the agarose polymer; form III corresponds to the linear conformation that is generated from the cleavage of form II, and this form is much more mobile than form II [15,34].
For compounds L1, L2, and complexes 3 and 4 (see Fig. 9a, Fig. 9b, Fig. 9c and Fig. 9d) no differences in their oxidative cleavage activities compared to that of the peroxide control were observed (Lane 3); however, a slight decrease in intensity in the line that corresponds to form I is seen, which suggests a possible decrease in the amount of this form of the plasmid.
Furthermore, a concentration dependence in the oxidative cleavage by Eu(III) complexes 5 and 6 was observed, and the band corresponding to form II increased in intensity as the amount of complex increases (see , Fig. 9e and Fig. 9f, lanes 5 to 8), which means that the relaxation of the plasmid to form II is favored. This could be explained by comparing the redox properties of the three metal ions (La(III), Eu(III) and Nd(III)). Eu(III) possesses more stable redox states (it can go from 3+ to 2+ and vice versa) compared to La(III) and Nd(III), which can only stay in the 3+ oxidation state. This behavior leads to a strong interaction between the reactive oxygen species and the DNA strands [34].
Otherwise, a reduction in the intensity in the line corresponding to form II for complexes 3, 5 and 6 (see Fig. 9c, Fig. 9e and Fig. 9f, lane 4) with respect to the DNA control in lane 2 is observed. This indicates that the complexes convert the plasmid from form II to form I as a consequence of induced rolling.
To elucidate the role of reactive oxygen species (ROS) in the cleavage of DNA pBR322 induced by the complexes, a preliminary study was conducted that suggested that H2O2 and the hydroxyl radical scavenger (DMSO) have little effect on the scission of DNA. When the experiment was carried out in the presence of KI, the content of form I was markedly reduced. This suggests that the species derived from hydrogen peroxide are those that actually take part in the DNA cleavage, in the experiments carried out using the Eu(HI) complexes, the active participation of the complexes in the conversion of form II to I was observed, which confirmed the results obtained in the previous assays.
The interaction modes observed in all the assays could be possible pathways for cytotoxic activity against a range of microorganisms. DNA binding could lead to interruption of the protein transcription mechanisms, which could be very useful against tumoral cells as well [28,29].
Antibacterial assays
Table 2 presents the MICs determined by the microdilution assays for the selected compounds against Staphylococcus Aureus (ATCC 25923).

The antibacterial activities of the compounds were lower for gram-negative bacteria (ATCC 25922 (Escherichia coli), ATCC 14028 (Salmonella Typhimurium), ATCC 2146 and ATCC 1705 (Klebsiella pneumoniae) over the evaluated concentration range; the obtained MICs were 2000 μg-mL -1 or higher. However, for the gram-positive strain ATCC 25923 (Staphylococcus aureus), complex 6 was found to have a MIC of 63 pg-mL -1 and major bactericidal behavior was observed in general. This result suggests that the cytotoxicity activity against bacteria may be dependent on the type of cell wall. Additionally, based on the MICs obtained for the ligands and the complexes, the biological activity was enhanced by the presence of the metal center.
According to the experimental results obtained in this study, the antimicrobial activity of the compounds may be influenced by the partial release of small and hard Eu(III) ions that can more efficiently affect the biosynthesis of peptidoglycan in the cell wall of the gram-positive bacteria, causing structural modifications and subsequent cell death. In addition to the conductivity, solubility and dipole moment, which are affected by the presence of metal ions, another possible mechanism that can explain the antimicrobial activity is chelation; and the coordination of thiosemicarbazones to the Lanthanide ions improves the lipophilicity of the complexes, thus favoring their penetration into the lipid membrane of the cell wall. In general, the complexes can inhibit a series of enzymes that play important roles in different metabolic processes that affect the development and growth of microorganisms. This is a similar result to those found in the DNA interaction assays, where the Eu(III) complexes were expected to have substantial biological activities based on the strength of its interaction with the strands.
Conclusions
To evaluate the potential biological activities that novel molecules could present in the fight against antimicrobial resistance and the discovery of new disease treatments, six new lanthanide complexes of La(III), Eu(III) and Nd(III) with N (4)-substituted thiosemicarbazones derived from the 2-carboxibenzaldehyde were synthesized to evaluate their interaction modes with DNA strands. The syntheses of 2-carboxybenzaldehyde N(4)-phenylthiosemicarbazone and 2-carboxybenzaldehyde N(4)-(4-chlorophenyl) thiosemicarbazone were carried out by preparing the corresponding thiosemicarbazides and condensing the subsequent Schiff base with 2-carboxybenzaldehyde. This was an efficient route for obtaining high-pure ligands with yields over 90 %. The lanthanide complexes were also obtained in high yields (over 80 %) and high purity. Additionally, they showed 1:2 (metal to ligand) stoichiometry in which metal ions are coordinated to the thiocarbonyl (C=S), azomethine (C=N) and carboxylate (COO-) groups (with the carboxylate serving as a bidentate ligand) according to IR, 1H-NMR and 13C-NMR spectral analyses. The low molar conductivities corroborated the non-electrolytic nature of the complexes and the possible coordination of one chlorine atom.
According to the results obtained from the viscosity measurements, electronic spectra and oxidative cleavage reactions, complexes 3, 5 and 6 can interact with the DNA strands through the phosphate backbone and promote bending or rolling of the plasmids. Furthermore, the Eu(HI) complexes catalyzed the oxidative cleavage of the DNA under high-oxidative-stress conditions. The oxidative cleavage by the complexes was concentration-dependent; the amount of complex increased the amount of cleavage to form II. This behavior was attributed to the redox properties of the Eu(III) ion. The antibacterial activity assays showed that the complexes possess bactericidal properties against the gram-positive strain ATCC 25923 (Staphylococcus Aureus) with MICs up to 63 μg-mL -1 for complex 6. Additionally, a cell-wall dependence was found in the biological activity of the lanthanide complexes; they may be more active against gram-positive bacteria. A synergistic effect of the metal center was also observed as higher inhibitions were obtained with the Eu(III) complexes compared to those observed with the ligands.

