











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
La malaria es una patología causada por parásitos del género Plasmodium, transmitidos al humano por la picadura de mosquitos hembra del género Anopheles, vectores del parásito. Existen cinco especies de Plasmodium causantes de malaria en humanos, dos de estas, Plasmodium falciparum y Plasmodium vivax, son las responsables de la mayor carga de la enfermedad asociada con esta infección.
Según el reporte mundial para la malaria, durante el 2018 se estimaron 228 millones de casos y 405.000 muertes en todo el mundo. La especie más prevalente en África es P. falciparum, causante de la mayor tasa de mortalidad por malaria en el mundo. Por otro lado, P. vivax es la especie predominante en países fuera de África, fundamentalmente en Asia y América (WHO, 2019). Durante el 2019 se notificaron en Colombia 78.513 casos, con un predominio de P. falciparum (50,3% de los casos), seguido de P. vivax (48,6%) e infección mixta (1,1%) (Instituto Nacional de Salud, 2019). Para el periodo epidemiológico VII, comprendido entre junio y julio de 2020, se notificaron 43098 casos de malaria (INS, 2020).
Convencionalmente, el diagnóstico de esta infección se realiza mediante el análisis microscópico de la gota gruesa y el extendido delgado de sangre periférica. Estos métodos poseen un límite de detección que puede variar en función de la experticia del observador, con un promedio de 5-10 parásitos/uL de sangre para un analista experimentado, y 20 a 25 parásitos para analistas menos experimentados (Nazaré et al, 2014). Factores como la calidad del proceso de recolección, el extendido, la tinción de la muestra de sangre y la pericia en la observación microscópica pueden generar falsos negativos, los cuales generan un mal diagnóstico clínico y la necesidad de repetir la prueba (Berzosa et al, 2018). Por otro lado, existen pruebas de diagnóstico rápido, basadas en el principio de la inmunocromatografía, para detectar antígenos derivados de Plasmodium spp. en la sangre humana (Cortés y Guerra, 2020). Dos de los antígenos más empleados son la proteína lactato deshidrogenasa (pLDH) (Vásquez et al, 2018), producida por todos los parásitos del género Plasmodium y la proteína rica en Histidina II (HRP-II) (Pava, Murillo, Echeverry, y Díaz, 2010), producida por los trofozoítos y gametocitos jóvenes de P. falciparum. La HRP-II puede permanecer en circulación hasta 14 días después de administrar un tratamiento antimalárico, razón por la cual puede ocasionar resultados falsos positivos durante este periodo de tiempo; sin embargo, por su practicidad, pueden apoyar el diagnóstico en situaciones donde no hay acceso a otros métodos diagnósticos (Gamboa et al, 2010; Pava et al, 2010; Vásquez et al, 2018).
Por lo anterior, es crucial identificar adecuadamente la especie de Plasmodium spp. y cuantificar la parasitemia en el paciente, para realizar un tratamiento correcto y oportuno y prevenir el desarrollo de posibles complicaciones fatales a causa del aumento en la parasitemia.
Con el desarrollo de pruebas moleculares como la PCR (Polymerase Chain Reaction), hoy en día usada principalmente en el contexto de la investigación, se logran confirmar los resultados obtenidos con los métodos convencionales de diagnóstico; además, son especialmente útiles cuando se presentan infecciones maláricas con parasitemias muy bajas, no detectadas por la microscopía o pruebas rápidas. Una PCR convencional usada para el diagnóstico de la malaria logra detectar hasta 1 parásito/μL, con una especificidad y sensibilidad cercanas al 100% (Grignard et al, 2020; Kassaza et al, 2018; Snounou y White, 2004). Al presentar mayor sensibilidad y especificidad que una gota gruesa, la PCR permite la diferenciación de las especies de Plasmodium que parasitan al hombre, incluso cuando se presentan bajas densidades parasitarias e infecciones submicroscópicas (Montiel et al, 2020a; Montiel et al, 2020b). En Colombia, la prevalencia de infecciones submicroscópicas oscila entre 4% y 12% en regiones con diferentes endemicidades (Vallejo et al, 2015). Otra gran ventaja de la PCR es que logra detectar infecciones mixtas. Se han descrito en la literatura protocolos de PCR multiplex en tiempo real que logran este propósito, cuya sensibilidad y especificidad son del 100% (Johnston et al, 2006; Lau et al, 2015).
Una variante de la PCR convencional es la PCR anidada (nested PCR-nPCR); en esta metodología primero se realiza una reacción con cebadores externos para amplificar una primera región de ADN más extensa, que contiene el segmento de interés. Después, este producto de amplificación se utiliza como molde para una segunda PCR en la cual se usan cebadores internos para amplificar una región más específica. Para la nPCR en malaria, la primera PCR genera un producto que permite identificar el género Plasmodium, y la segunda reacción permite amplificar una secuencia específica de especie, al tiempo que se incrementa la sensibilidad de esta herramienta molecular (Mfuh et al, 2019; Pöschl, Thekisoe, Chutipongvivate, Panagiotis, y Waneesorn, 2010). Una desventaja de la alta eficiencia de esta amplificación es que la nPCR se vuelve intrínsecamente más susceptible a una reamplificación de amplicones generados en reacciones previas, dando como resultado una contaminación por amplicones (Longo, Berninger, y Hartley, 1990). La contaminación de las reacciones de PCR con ADN exógeno es uno de los problemas más comunes que afectan la detección de ácidos nucleicos, generando falsos positivos o incluso la inhibición de las reacciones (Bacich, Sobek, Cummings, Atwood, y O'Keefe, 2011; Borst, Box, y Fluit, 2004). La contaminación por arrastre o contaminación con amplicones, que contienen secuencias blanco, puede ocurrir frecuentemente en los laboratorios donde se procesan constantemente muestras para PCR y el remanente de las amplificaciones puede conducir a un diagnóstico erróneo (Bacich et al, 2011; Borst et al, 2004). En este contexto, los amplicones generados podrían contaminar los reactivos, las pipetas, las superficies del espacio de trabajo o inclusive la piel de los analistas. Para prevenir el problema de la contaminación se han descrito diversas estrategias, algunas aprovechan los efectos de agentes físicos como la luz ultravioleta, hasta agentes químicos que degradan la doble hélice de ADN (Aslanzadeh, 2004), entre otras metodologías. Una de estas estrategias, de tipo enzimático, se basa en el uso de la enzima uracilo ADN glicosilasa (UDG) (EC:3.2.2.27) y puede incorporarse directamente en la mezcla de reacción para la PCR, (Fallahi et al, 2018; Hsieh, Mage, Csordas, Eisenstein, y Soh, 2014; Longo et al, 1990; Wang, Chen, Kim, y Piomelli, 1992).
La uracilo-ADN glicosilasa es una enzima con actividad exonucleasa, presente en casi todos los organismos vivos y su función es la reparación del ADN por escisión de bases. Actúa reparando mutaciones que se generan en el ADN, como la oxidación de citosinas que se convierten en deoxi-uracilos (Aslanzadeh, 2004). La enzima UDG genera una hidrólisis del enlace N-glicosídico, que une la base nitrogenada uracilo con el esqueleto de azúcar-fosfato, en la doble hélice del ADN (Schormann, Ricciardi, y Chattopadhyay, 2014). En su estado natural, el ADN tiene incorporado el nucleótido timina y no uracilo, sin embargo, si se utilizan uracilos o dUTPs en lugar de timinas en una reacción de PCR, la taq polimerasa logra amplificar con igual eficiencia la doble cadena de ADN (Bessman et al, 1958; Mosbaugh, 1988). La enzima UDG es capaz de degradar los nucleótidos de uracilo contenidos en un producto de amplificación de PCR; de esta manera, puede establecerse un sistema in vitro, en el que se realiza la amplificación del ADN blanco incorporando uracilo en lugar de timina, y se adiciona UDG a la mezcla de reacción de nuevas muestras, la cual se espera que degrade amplicones generados en otras reacciones que contienen uracilo y que puedan estar contaminando la reacción actual, sin afectar el ADN nativo de la nueva reacción, que contiene timina y no uracilo. Mediante esta estrategia, Longo et al (1990), incorporaron por primera vez esta enzima en una RT-PCR para la detección del virus del papiloma humano, con el objetivo de que cualquier ADN amplificado en reacciones previas fuese degradado, si llegase a contaminar los reactivos o las mezclas de una nueva reacción (Longo et al, 1990).
Varios investigadores han empleado la UDG en sus protocolos PCR, para diferentes aplicaciones, como en la amplificación de genes de hemoglobina humana (Wang et al, 1992), en una RT-PCR para la detección de coxsackievirus (Pang, Modlin, y Yolken, 1992), y más recientemente, en una PCR para la detección de Toxoplasma gondii (Martins et al, 2000), convirtiéndose en el primer reporte del uso de la enzima para una PCR cuyo blanco es ADN parasitario.
En los últimos años este sistema se ha incorporado en diferentes kits comerciales de PCR en tiempo real (qPCR); un ejemplo es el Platinum™ PCR SuperMix de Invitrogen. Estas marcas comerciales incorporan en la mezcla de reacción la UDG y los dUTPs, para usarlos sin necesidad de añadirlos de manera manual en las mezclas de reacción de PCR; no obstante, el costo de estos kits puede ser una limitante para laboratorios con recursos limitados. El objetivo de este trabajo fue diseñar una estrategia para la incorporación del sistema UDG-dUTPs en reacciones de nPCR para la detección de P. falciparum y P. vivax, como una alternativa para reducir el riesgo de contaminación con amplicones que podría generar resultados falsos positivos, en esta prueba molecular utilizada para confirmar el diagnóstico de la malaria.
MATERIALES Y MÉTODOS
ADN de P. falciparum y P. vivax. La fuente de ADN fue la cepa 3D7 de P. falciparum, cultivada en el laboratorio del Grupo Malaria de la Facultad de Medicina de la Universidad de Antioquia y dos muestras de pacientes con diagnóstico de malaria por P. vivax confirmadas por PCR. La extracción se realizó mediante el kit comercial QIAamp DNA Blood Mini (QIAgen 51106). Los eluidos de extracción de ADN se cuantificaron mediante fluorescencia utilizando el kit comercial de cuantificación de ADN (ref. Q32851) del equipo Qubit (Thermofisher); luego, fueron distribuidos en alícuotas almacenadas a -20 °C para realizar los distintos experimentos del estudio.
PCR anidada para la detección de Plasmodium spp. Para la nPCR se adaptaron los cebadores que amplifican un fragmento de 1200 pares de bases, común para el género Plasmodium; a partir de este fragmento se amplificaron secuencias de los genes de la subunidad ribosomal 16s tanto para P. falciparum como para P. vivax, siguiendo lo reportado por Singh et al (1999).
La mezcla de PCR para amplificar el primer fragmento, correspondiente al género Plasmodium, se preparó en un volumen final de 20 µl, añadiendo 2 µl de Buffer 10X (NH4)2SO4-MgCl2 (Thermo Scientific, M8295), 1,6 µl de MgCl2 25 mM (Thermo Scientific, M8295), 2 µl de dNTPs (dATP, dCTP, dGTP, dTTP), 10 mM cada dNTP (Fermentas, R0193), 0,5 µl de cebador rPLU1 10 µM 5´ TCA AAG ATT AAG CCA TGC AAG TGA 3´ (Macrogen), 0,5 µl de cebador rPLU5 10 µM 5´ CCT GTT GTT GCC TTA AAC TTC 3´ (Macrogen), 0,4 µl de Taq Polimerasa a 2 U/PCR (Thermo), 11 µl de agua PCR (Amresco, E476-500 mL), y finalmente, 2 µl de muestra de ADN. La PCR se corrió en el termociclador Biometra T3. La amplificación se realizó durante 4 min a 94 °C, 35 ciclos de 94 °C por 30 s, 55 °C por un min y 72 °C por un min y una incubación final a 72°C por 4 min. El producto amplificado, a partir de la reacción de género, fue usado como molde para la amplificación del fragmento de especie. Para cada reacción de especie se emplearon los cebadores publicados por Singh et al (1999), específicos de secuencias que codifican ARN de la subunidad ribosomal 16S de cada una de las especies: P. falciparum y P. vivax; la mezcla de la reacción se realizó añadiendo 2 µl de Buffer 10X (NH4)2SO4-MgCl2 (Thermo Scientific, M8295), 1,6 µl de MgCl2 25 mM (Thermo Scientific, M8295), 2 µl de dNTPs (dATP, dCTP, dGTP, dTTP), 10 mM cada dNTP (Fermentas, R0193), 0,5 µl cebador rFAL1 10 µM 5´-TTA AAC TGG TTT GGG AAA ACC AAA TAT ATT -3´ (Macrogen) y 0,5 µl rFAL2 10µM 5´- ACA CAA TGA ACT CAA TCA TGA CTA CCC GTC-3´ (Macrogen), 0,1 µl de Taq polimerasa 5 U/μL, para un volumen final de 20 µl al incorporar 2 µl del primer producto amplificado obtenido en la reacción de género. Las mismas concentraciones de reactivos fueron utilizadas para la mezcla de reacción de P. vivax, con la adición de 0,8 µl de dimetilsufóxido al 100% a la mezcla de reacción y empleando 0,5 µl de cada uno los cebadores específicos para P. vivax: rVIV1 5´- CGC TTC TAG CTT AAT CCA CAT AAC TGA TAC-3´(Macrogen) y rVIV2 5´- ACT TCC AAG CCG AAG CAA AGA AAG TCC TTA-3´ (Macrogen). La amplificación se realizó en el termociclador Biometra T3, a 94 °C por un min, 35 ciclos con las temperaturas de 94 °C por 30 s, 62 °C por un min y 72 °C por un min; finalmente, una incubación a 72 °C por 4 min. El producto de amplificación, de 240 pb para P. falciparum y 117 pb para P. vivax, fue visualizado en gel de agarosa al 1% teñido con gel red (Biotium ref.41003).
Sistema UDG-dUTPS. Para la la incorporación de la enzima UDG y los nucleótidos dUTPs, fue necesario realizar una modificación de la mezcla de reacción y de la solución de trabajo de nucleótidos. El primer paso fue sustituir la timina por uracilo en todas las reacciones anidadas, así la mezcla de dNTPs fue preparada con dUTPs (Thermo scientific, R0133) y el resto de los nucleótidos dATPs, dCTPs, dGTPs en concentraciones iguales de cada uno, para obtener una solución de trabajo a una concentración de 2 mM. De este modo se aseguró que todos los productos de amplificación futuros en la reacción anidada contendrán uracilo.
Para evidenciar la actividad degradante de la enzima UDG sobre los productos amplificados con uracilo (amplicones contaminantes), se modificó la mezcla de reacción para la PCR de especie, adicionándole una U/μL de UDG. El volumen final de la reacción fue 20 μL, con 2 µl de Buffer 10X (NH4)2SO4-MgCl2 (Thermo Scientific, M8295), 1,6 µl de MgCl2 25mM (Thermo Scientific, M8295), 2 μL de dNTPs (dATP, dCTP, dGTP, dUTP) a 2 mM cada dNTP (Fermentas, R0193), 0,1 µl de Taq polimerasa 5 U/μL, 0,5 μL de cebador rFAL1 (Macrogen) y 0,5 μL de cebador rFAL2 (Macrogen) para P. falciparum o 0,5 μL de cebador rVIV1 (Macrogen) y 0,5 μL de cebador rVIV2 (Macrogen) con 0,8 µl de Dimetilsufóxido al 100% para P. vivax, ajustando el volumen de cada reacción con agua PCR y 1 μL de UDG (Thermo Scientific, EN0361). Siguiendo las instrucciones del fabricante, se realizó una incubación a 37 °C por 15 min, condiciones en las que la UDG funciona óptimamente, y luego una inactivación enzimática a 95 °C por 10 min. Posterior a las incubaciones, se llevó a cabo la amplificación a 94 °C por un min, luego 35 ciclos a 94 °C por 30 s, 62 °C por un min y 72 °C por un min; finalmente, una incubación a 72 °C por 4 min.
Límite de detección de la nPCR. Se calculó el límite de detección de la nPCR convencional y la nPCR con dUTPs. Para esto, se usó una serie de controles obtenidos a partir de un cultivo sincronizado, con un 80% de formas parasitarias, en estado de anillos, de la cepa 3D7 de P. falciparu; se realizaron diluciones seriadas de este cultivo utilizando glóbulos rojos humanos sanos, obteniendo así diluciones entre 1000 y 0,1 parásitos/μL. La parasitemia en cada dilución del cultivo fue cuantificada mediante gota gruesa, con base en el recuento de 200 leucocitos. De cada una de estas diluciones se hizo extracción de ADN utilizando el Kit QIAamp DNA Blood Mini (Qiagen cat. 51106); se utilizaron 2 μL de ADN de cada dilución para cada reacción de nPCR. La eficiencia de las extracciones de ADN se verificó mediante la cuantificación con el equipo nanodrop 2000c Spectrophotometer (Thermo Scientific); las lecturas de diluciones de 912,5 parásitos/μL, 91,35 parásitos/μL, 9,125 parásitos/μL, 0,913 parásitos/μL y 0,091 parásitos/μL, fueron correspondientes a concentraciones de ADN de 5,2 ng/μL, 1,9 ng/μL 1,6 ng/μL 1,1 ng/μL 0,8 ng/μL respectivamente. Para el caso de P. vivax se realizaron diluciones seriadas de la elución del ADN extraído a partir de una muestra clínica confirmada por PCR.
Análisis de resultados. Se establecieron comparaciones cualitativas, mediante el análisis de los productos de amplificación en geles de electroforesis, de la reacción de nPCR original y aquella incorporando el sistema UDG-dUTPs. Cada experimento se realizó por triplicado.
RESULTADOS
El límite de detección de la nPCR no se afectó cuando se reemplazó la timina por uracilo.
Para comprobar que la Taq polimerasa es igualmente eficiente en el proceso de elongación de la cadena de ADN, en presencia de nucleótidos de timina (dTTPs) y de uracilo (dUTPs), se ejecutó la nPCR utilizando mezclas de reacción para la amplificación del fragmento de especie con timina o con uracilo. Cabe aclarar que la sustitución de los nucleótidos de timina por uracilo solo se hizo en las mezclas de reacción para la segunda ronda de amplificación o anidadas, ya que, de lo contrario, si este cambio se realizara desde la primera amplificación, todo el ADN amplificado sería degradado en la subsiguiente reacción puesto que en las mezclas de reacción anidadas se incorpora la enzima UDG.
La nPCR funcionó correctamente para amplificar el blanco molecular de especie para P. falciparum (fragmento de 240 pb) como para P. vivax (fragmento de 117pb) tanto con dTTPs como con dUTPs, pues se observaron bandas de similar intensidad en los geles de electroforesis usados para revelar el producto de ambas reacciones (Figuras 1 y 2).

Figura 1 nPCR para P. falciparum usando A. dTTPs y B. dUTPs. 1) Control negativo. 2) Control positivo. 3) Dilución de cultivo 3D7 correspondiente a 912,5 parásitos/μl. 4) 91,35 parásitos/μl. 5) 9,125 parásitos/μl. 6) 0,913 parásitos/μl. 7) 0,091 parásitos/μl. 8) Marcador de peso molecular.
Bajo esta premisa, con el ADN proveniente de las diluciones seriadas de cultivo 3D7, se prepararon reacciones para calcular el límite de detección en la nPCR para P. falciparum, el cual fue de 0,091 parásitos/μL con dTTPs, es decir con la mezcla de reacción estándar; en contraste, se observó una ligera disminución en la sensibilidad de la nPCR cuando se usaron dUTPs, ya que para esta se observó una banda franca en el gel de electroforesis solo hasta la dilución correspondiente a 0,913 parásitos/μL (Figura 1).
Para P. vivax el límite de detección se realizó utilizando diluciones seriadas del ADN de sangre total previamente extraído de una muestra de paciente con infección por P. vivax, cuya parasitemia fue 33.000 parásitos/μL; con estos datos se ajustaron, teóricamente, las concentraciones para obtener desde 1.000 parásitos/ μL hasta 0,1 parásitos/ μL. Para cada reacción de nPCR se emplearon 2 μL de cada dilución usando los protocolos de mezclas de reacción con dTTPs y dUTP. El límite de detección obtenido fue 10 parásitos/μL para ambos protocolos (Figura 2).

Figura 2 nPCR para P. vivax usando dTTPs y dUTPs. 1) Marcador de peso molecular. 2) Control negativo. 3) Control positivo. 4) 1000 parásitos/μl. 5) 100 parásitos/μl. 6) 10 parásitos /μl. 7) 1 parásito/μl. 8) 0,1 parásitos/μl. 9) 1000 parásitos/μl. 10) 100 parásitos/μl. 11) 10 parásitos /μl. 12) 1 parásito/μl. 13) 0,1 parásitos/μl. 14) Marcador de peso molecular.
Efecto de la enzima UDG en reacciones contaminadas artificialmente con amplicones. Para comprobar el efecto catalítico de la UDG sobre los productos amplificados con uracilo (contaminante), se realizó un experimento en el cual, mezclas de reacción para la nPCR para P. falciparum (controles negativos) fueron contaminados artificialmente con un producto amplificado con uracilo correspondiente a fragmentos específicos de P. falciparum y cuya concentración de ADN, medida por nanodrop, fue 580 pg/μl. Estas mezclas de reacción contaminadas fueron incubadas con o sin la adición de 1 U/μl de la enzima UDG durante 15 min a 37 °C. Con el objetivo de comprobar un posible efecto inhibidor de la reacción de amplificación cuando esta es realmente positiva, se incluyó una mezcla de reacción con ADN nativo (control positivo) para P. falciparum, a la cual se indujo la contaminación descrita anteriormente. La enzima UDG logró eliminar totalmente el producto contaminante de 580 pg en la reacción de nPCR para la detección de P. falciparum. Además, no se encontró ninguna interferencia y la amplificación fue exitosa, inclusive cuando se contaminaron las mezclas de reacción en los controles positivos correspondientes al ADN nativo (Figura 3).
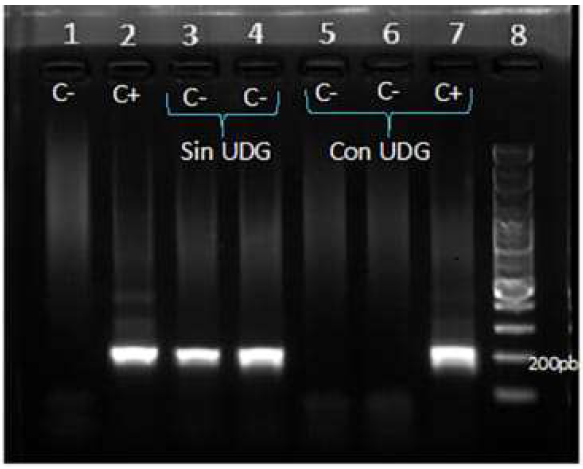
Figura 3 Efecto de la enzima UDG en la reacción de nPCR para P. falciparum contaminada artificialmente con amplicones. 1) Control negativo sin contaminación y sin UDG. 2) Control positivo sin contaminación y sin UDG. 3-4). Controles negativos contaminados con 580 pg de producto de amplificado con dUTP. 5-6) Controles negativos contaminados con 580 pg de producto de amplificación con dUTPs e incubados con 1 U/μl de enzima UDG. 7) Control positivo contaminado con 580 pg de producto de amplificación con dUTPs e incubados con 1 U/μl de enzima UDG. 8) marcador de peso molecular.
Límite de degradación de la UDG en reacciones contaminadas. Para comprobar que la presencia de contaminantes en la PCR puede generar falsos positivos y que estos pueden ser eliminados a cabalidad por la enzima UDG, se corrieron controles negativos, previamente contaminados con 1 μL de producto amplificado con dUTPs con cebadores específicos para la especie P. falciparum, para cubrir un rango de concentraciones contaminantes de 5,8 ng, 580 pg, 58 pg, 5,8 pg, 0,58 pg y 0,058 pg en la mezcla de reacción. Para determinar el límite de degradación de la UDG en la nPCR para P. vivax se utilizó un producto amplificado con dUTPs como contaminante en diluciones de 4,9 ng, 490 pg, 49 pg, 4,9 pg, 0,49 pg y 0,049 pg. De cada dilución se añadió 1 μL en las mezclas de reacción de nPCR específicas para P. vivax. Para cada caso, las nPCRs fueron incubadas con o sin adición de UDG (1 U/μL) en sus mezclas de reacción.
Se evidenció que 1 U/μL de UDG puede eliminar, efectivamente, los productos contaminantes en su totalidad, en cargas de hasta 58 y 49 pg y puede degradar, parcialmente, una masa de producto contaminante de aproximadamente 500 pg y 5 ng para la PCR anidad de P. falciparum (Figura 4) y P. vivax (Figuras 5).

Figura 4 Límite de degradación de la enzima UDG en reacciones de nPCR para P. falciparum, contaminadas con ADN conteniendo uracilo. 1) y 16) Marcador de peso molecular. 2) Control negativo. 3) Control positivo. 4-9) Controles negativos contaminados con 5,8ng, 580pg, 58pg, 5,8pg, 0,58pg y 0,058pg de amplicones con dUTPs respectivamente. 10-15). Controles negativos contaminados con 5,8 ng, 580pg, 58pg, 5,8pg, 580fg y 58fg de amplicones con dUTPs e incubados con 1 U/μL de enzima UDG durante 15 min a 37 °C.

Figura 5 Límite de degradación de la enzima UDG en reacciones de nPCR para P. vivax, contaminadas con ADN conteniendo uracilo. 1) y 16) Marcador de peso molecular. 2) Control negativo. 3) Control positivo. 4-9). Controles negativos contaminados con 4,9 ng, 490pg, 49pg, 4,9pg, 0,49pg y 0,049pg de amplicones con dUTPs respectivamente. 10-15) Controles negativos contaminados con 4,9 ng, 490pg, 49pg, 4,9pg, 0,49pg y 0,049pg de amplicones contaminantes e incubados con 1 U/μl de enzima UDG durante 15 min a 37 °C.
DISCUSIÓN
Con el objetivo de mejorar la calidad de los resultados de la nPCR, en el modelo de P. falciparum y P. vivax, se incorporó el sistema UDG-dUTPs en esta reacción para diseñar una estrategia metodológica que evite la contaminación por arrastre con productos previamente amplificados.
Martins et al (2000) registraron, por primera vez, el uso de este sistema para una PCR cuyo blanco molecular fue un ADN parasitario. Hasta donde conocemos, este es el primer registro de la incorporación “in house” del sistema UDG-dUTPs para el diagnóstico de P. falciparum y P. vivax en una nPCR, aportando al avance de los métodos moleculares utilizados en rutina en investigación y en la confirmación del diagnóstico de la malaria.
A pesar de la disponibilidad de diferentes métodos para mitigar la contaminación por arrastre o por amplicones, la estrategia de utilizar la UDG es completamente distinta a cualquier otra, ya que esta enzima actúa directamente sobre las mezclas de reacción sin afectar o degradar los componentes de estas. El uso de la UDG no excluye o descarta la implementación de otros métodos para evitar la contaminación (Aslanzadeh, 2004) como el uso de la luz UV, el hipoclorito de sodio, el acondicionamiento del espacio físico de trabajo, el direccionamiento del flujo de aire, entre muchas otras estrategias que son efectivas para eliminar los contaminantes o amplicones de las superficies, equipos e instrumentos. Sin embargo, ninguno de estos métodos, usado individualmente, es efectivo en un 100% para eliminar dichos contaminantes de las reacciones de amplificación que se llevan a cabo en laboratorios de investigación o de diagnóstico, por lo cual reviste gran importancia el uso de métodos enzimáticos, como la UDG, que actúen directamente sobre las reacciones de amplificación de ADN.
En cuanto a la incorporación de uracilo, confirmamos que los dTTPs pueden ser sustituidos por dUTPs y que la Taq polimerasa amplifica, con igual eficiencia, el blanco molecular de interés. A diferencia de otros modelos de PCR que han incorporado la UDG, en este estudio no se modificó la concentración de uracilos (dUTPs), con respecto a la concentración convencionalmente usada de los nucleótidos con timina (dTTPs a 2 mM). En nuestros experimentos se logró, sin inconvenientes, la amplificación de ADN con uracilo, a pesar que otros autores reportan la necesidad de ajustar e incrementar la concentración de dUTPs y MgCl2. Al respecto,Pang et al (1992) aumentaron la concentración estándar de dUTPs de 200 μM a 600 μM y la cantidad de MgCl2 de 1 mM a 2 mM; Wang et al (1992), registraron en sus ensayos un aumento de la concentración de dUTPs de 200 μM hasta 1.000 μM, y el incremento de 1.5 mM hasta 5 mM de MgCl2, concentraciones de MgCl2 con las cuales se obtienen mejores resultados en cuanto a la amplificación obtenida con sus protocolos de PCR.
Wang et al (1992), discuten que el incremento de la concentración de dUTPs y MgCl2 mejoró la fidelidad de la amplificación en la PCR. Las evidencias de Pang et al (1992), confirman que el incremento en las concentraciones de estos reactivos permite que la amplificación sea más efectiva y, por ende, se obtienen bandas de gran intensidad en los geles de electroforesis. A pesar de estas afirmaciones es probable que, debido a que el tipo de ADN utilizado en sus experimentos, el cual fue ADN copia a partir de extracciones de ARN de coxsakievirus y ADN humano, matrices distintas a las usadas en nuestros experimentos, los cuales se realizaron a partir de ADN total, el cual contiene tanto ADN parasitario como ADN humano, exista una leve interferencia en la amplificación del ADN plasmodial en la nPCR.
La nPCR presentó un buen desempeño en cuanto al límite de detección de P. falciparum, comparado con los resultados obtenidos por Singh et al (1999), quienes reportaron una PCR positiva con 6 parásitos/μL de sangre. En nuestro laboratorio, el límite de detección obtenido fue 0,1 parásitos/μL aproximadamente y se observó una ligera disminución en la sensibilidad del método cuando se incorporó uracilo en las reacciones. Frente a esto es posible concluir que la PCR posee una alta sensibilidad analítica a pesar de sustituir dTTPs por dUTPs en las reacciones de especie, ya que la Taq polimerasa puede sintetizar cadenas de ADN con igual eficiencia con timina y uracilo, al no discriminar entre estos nucleótidos.
La lógica para poder incorporar el sistema de UDG-dUTPs, implicó que sólo se podrían sustituir los dTTPs por dUTPs en las reacciones de especie y no en las reacciones de género. Esto debido a que, si se amplificaran productos de género con uracilo y luego fuesen preincubados con UDG en la reacción anidada o de especie, se degradarían en su totalidad, generando falsos negativos en las reacciones al degradarse todo el ADN amplificado con uracilo; esto no ocurre cuando se amplifican fragmentos de género con timina y se preincuban dichos productos amplificados de género en las reacciones de especie con UDG puesto que en este escenario no se degradaría el ADN previamente amplificado. Por lo anterior, sólo productos de especie se amplificaron con uracilo, a pesar que una reacción de nPCR podría contaminarse desde la mezcla de género; sin embargo, los cebadores de la PCR de género al no coincidir con los amplicones contaminantes generados en las reacciones de especie, no podrían amplificar este material contaminante en la primera ronda de amplificación y este contaminante sólo podrá manifestarse en las reacciones de especie en donde realmente podría ser reamplificado con éxito, generando falsos positivos. Esta estrategia se convierte en una desventaja del sistema UDG-dUTPs para una nPCR ya que solo es posible controlar la contaminación en un punto del proceso y no en los dos pasos de amplificación; por ende, el uso de la UDG limita y condiciona en qué punto se puede reemplazar la timina por uracilo, a diferencia de una reacción que no requiera ser de tipo anidada (Longo et al, 1990; Martins et al, 2000; Pang et al, 1992; Wang et al, 1992).
En la nPCR, para detectar P. falciparum se pueden generar falsos positivos cuando se induce una contaminación artificial con hasta 5,8 pg de amplicones, en cuanto a P. vivax, son necesarios 4,9 pg de amplicones para generar dicha contaminación; esta masa de producto contaminante es similar a la registrada por Longo et al (1990), en la cual se generan falsos positivos en una PCR para amplificar un plásmido del virus del papiloma humano serotipo 16, contaminada con concentraciones de hasta 0,1 pg de amplicones (Longo et al, 1990). La eliminación de productos contaminantes por efecto de la UDG en nuestros experimentos fue efectiva. Una unidad/μL de enzima UDG fue capaz de degradar sin problema un producto contaminante de 580 pg, sin inhibir una reacción positiva contaminada con esta misma carga de amplicones. Basados en otros autores, como Longo et al (1990), los productos de amplificación pueden contaminar una reacción de PCR cuando se incorporan hasta 100 pg de este en una mezcla de reacción (Longo et al, 1990); para nuestro hallazgo, un producto contaminante de hasta 5 pg aprox. puede contaminar efectivamente la reacción de nPCR.
En el diagnóstico molecular de la malaria, como en muchas otras patologías, la contaminación por amplicones puede generar una gran cantidad de errores en las amplificaciones de rutina y este riesgo es aún mayor cuando se procesa un gran número de muestras en los laboratorios diariamente. Como estrategia para dar solución a este inconveniente, la implementación del sistema UDG-dUTPs se convierte en un excelente modelo para prevenir falsos positivos en las reacciones de PCR, incluso cuando las concentraciones de contaminantes son extremadamente bajas. A pesar que diferentes marcas comerciales han incorporado este sistema en kits comerciales listos para usar, incorporar la UDG en la PCR que se lleva a cabo en el laboratorio del grupo malaria, generó un protocolo que puede emplearse de forma rutinaria y que funciona exitosamente. Finalmente, el desarrollo o mejora tecnológica de una nPCR para detectar P. falciparum y P. vivax genera un aporte en el diagnóstico molecular dado que, al ser un método utilizado para confirmar el diagnóstico, al presentarse casos con muy baja densidad parasitaria, como en el caso del monitoreo de la resistencia a antimaláricos, genera resultados seguros y confiables, así como diagnósticos correctos con tratamientos oportunos.
Esperamos que, con estos hallazgos, esta estrategia de descontaminación se pueda implementar, no sólo en una nPCR para la detección de P. falciparum y P. vivax, sino también para detectar otras especies que parasitan a los humanos, como P. malarie y P. ovale. Además, pretendemos evaluar en el futuro el desempeño de la nPCR con el sistema UDG-dUTPs para el diagnóstico de la malaria como prueba diagnóstica en una población determinada.
Otra perspectiva, en el contexto de las herramientas moleculares para el diagnóstico de malaria, es implementar el sistema UDG-dUTPs en una reacción de amplificación isotérmica como LAMP (Loop mediathed isothermal amplification); esto sería una gran aplicación debido a que esta técnica, además que puede emplearse como prueba confirmatoria del diagnóstico en condiciones de campo (Lucchi, Ljolje, Silva-Flannery, y Udhayakumar, 2016; Lucchi, Ndiaye, Britton, y Udhayakumar, 2018; Vásquez et al, 2018), posee una alta capacidad de amplificación, lo que la hace más susceptible a la contaminación por arrastre de amplicones. Algunos autores ya han demostrado la efectividad del sistema UDG-dUTPs en reacciones LAMP, para controlar la contaminación en otros modelos de diagnóstico (Hsieh et al, 2014; Kil et al, 2015; Tang, Chen, y Diao, 2016), por lo que se esperaría lograr estandarizar este tipo de amplificación con dicha estrategia de descontaminación, para el diagnóstico de la malaria.

