











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
La descripción original de los EIM (errores innatos del metabolismo) se debe al trabajo de Sir Archibald Garrod, quien acuñó el concepto a principios del siglo XX, al integrar los patrones de herencia mendeliana con la manifestación familiar de la enfermedad metabólica 1. Garrod, a través de la descripción de la alcaptonuria, estableció el reconocimiento clínico de la herencia recesiva y la relación entre bioquímica y signos y síntomas específicos. Desarrolló, además, el concepto de individualidad bioquímica 2, en el cual se establece que el metabolismo de cada persona atañe a características particulares que influyen el proceso salud enfermedad 3. Estos conceptos, pioneros en su tiempo, constituyen la definición fundamental de los EIM, los cuales comprenden un grupo de trastornos heterogéneos cuya base molecular consiste en la presencia de mutaciones en genes que se expresan como enzimas, proteínas activadoras, cofactores de enzimas, proteínas de transporte, sistemas portadores o marcadores de reconocimiento metabólico 4. La alteración de la función enzimática conlleva a la acumulación anormal de un substrato bioquímico, a la producción anormal de metabolitos y a la reducción de un producto. En algunos casos el cambio estructural anormal de la proteína puede implicar el incremento de la actividad enzimática y exceso de producto 5-7. La mayoría de estas enfermedades se heredan a través de un mecanismo de herencia autosómico recesivo, lo que implica que ambos padres biológicos deben ser portadores de al menos una de las alteraciones genéticas (mutaciones), la cuales, al presentarse en estado homocigoto en uno de sus hijos, condicionan la insuficiencia enzimática que originará la enfermedad 8.
El presente artículo tiene como propósito describir una serie de recomendaciones clínicas, para alcanzar oportunamente el diagnóstico de las enfermedades metabólicas en el periodo neonatal, enfatizando en la presentación aguda y su asociación con tres manifestaciones clínicas de máxima relevancia, como son la acidosis metabólica, la hiperamonemia y la hipoglicemia, las cuales requieren manejo urgente en la unidad de cuidado intensivo neonatal (UCIN).
Presentación clínica
Las características clínicas de los EIM de presentación aguda son secundarias al efecto de toxicidad por acumulación del substrato, déficit energético por disminución del producto o por compromiso funcional de moléculas complejas 9. Sin embargo, muchas de ellas pueden ser inespecíficas clínicamente durante la edad neonatal, de tal modo que la historia del embarazo y los antecedentes familiares como abortos espontáneos, muerte súbita en un hermano, inmunodeficiencias, fallas en el crecimiento, malformaciones o consanguinidad de los padres, son factores de alarma que hacen sospechar un EIM 10,11.
Los neonatos a término, con antropometría adecuada para la edad gestacional, parto sin complicaciones y adaptación neonatal adecuada, que presentan súbitamente deterioro del estado general, en quienes se hayan descartado otras causas que expliquen el deterioro, como por ejemplo infección de sistema nervioso central (SNC), sepsis o pobre succión secundaria a alteraciones neurológicas no metabólicas, tienen indicación para realizar estudios de EIM. Las principales manifestaciones que hacen sospechar una EIM se resumen en la tabla 1.
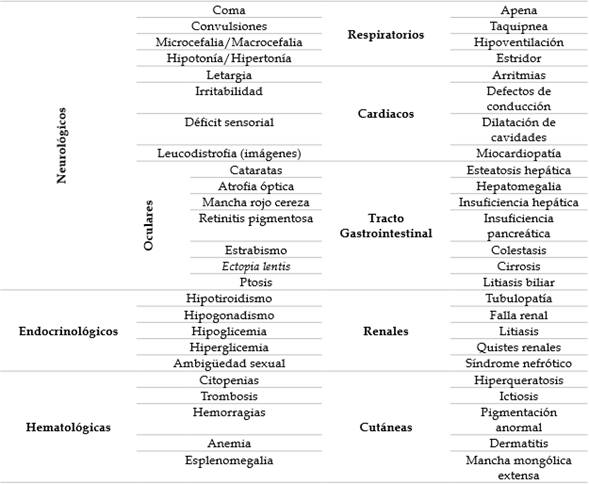
Las manifestaciones neurológicas pueden ser muy variadas y es usual que la sospecha de EIM se retrase hasta descartar otras etiologías. De tal modo que es preferible que los EIM deban considerarse desde un principio, en el trabajo diagnóstico de alteraciones neurológicas del neonato 12. El déficit sensorial, usualmente no se detecta de manera temprana en la UCIN, pero podría acompañar el cuadro tóxico, en un menor de un mes que reingresa a la unidad 13,14; sin embargo, es importante recalcar que las manifestaciones oculares como la catarata congénita, la mancha rojo cereza e incluso la retinitis pigmentosa, son aspectos que se deben investigar durante el examen físico completo del neonato, especialmente, si hay sospecha de EIM 15-17.
En relación a la encefalopatía aguda metabólica, esta es el resultado de la acción tóxica de metabolitos anormales en el SNC. Los principales hallazgos clínicos son letargia, pobre succión o intolerancia a la vía oral 18. El vómito es una característica importante de los EIM, especialmente el asociado a la intolerancia a la proteína, aunque es un signo de mayor relevancia en lactantes mayores y no en el neonato 19. Al descartar infección o sepsis, como diagnóstico diferencial, los EIM son la primera línea diagnostica; sin embargo, los pacientes afectados por EIM rápidamente pueden desarrollar sepsis, luego de cuadros infecciosos que de otra forma se resolverían, por tanto, el diagnóstico de patología infecciosa complicada no descarta el diagnóstico de EIM 20.
Si la causa de la letargia no es tratada rápidamente, esta progresa velozmente a coma y muerte 18,21. La letargia del paciente puede acompañarse de otros signos de disfunción de SNC como convulsiones, alteraciones del tono muscular, edema cerebral y hemorragias intracraneales 22. También se debe tener en cuenta que en relación al cuadro encefalopático, se presentan alteraciones del ritmo respiratorio, que incluso pueden ser el motivo de consulta, como son la apnea y la dificultad respiratoria 23. Las apneas de origen metabólico son típicamente de origen central, sin embargo la taquipnea puede ser un síntoma de la acidosis subyacente de una acidemia orgánica 24. Los pacientes con defectos del ciclo de la urea, presentan inicialmente hiperventilación central que conduce a alcalosis respiratoria 19,23. Las manifestaciones cardiacas también tienen un amplio rango de presentación, lo que incluye arritmias secundarias a la acumulación de metabolitos arritmogénicos 25, cardiomiopatía secundaria a déficit energético de las enfermedades mitocondriales o secundaria a trastornos del metabolismo de lípidos 26.
Las manifestaciones del tracto gastrointestinal abarcan desde la fibrosis hepática congénita, la hepatomegalia en las enfermedades de depósito y la disfunción hepática en los desórdenes del ciclo de la urea 27. La ictericia puede ser una manifestación de EIM y se manifiesta en la mayoría de los casos por una elevación de la bilirrubina directa 28 (excepto por los EIM relacionados con metabolismo del eritrocito que conllevan a hemolisis, como la deficiencia de Glucosa 6-fosfato deshidrogenasa o la deficiencia de piruvato quinasa) 29,30. El más conocido de los diagnósticos de EIM relacionado con ictericia es la galactosemia, en la que se genera toxicidad hepática por el galactitol y de galactosa 1-fosfato. La ictericia de la galactosemia se asocia a falla hepática progresiva, alrededor de la segunda semana de vida y se asocia a vómito, diarrea, pobre ganancia de peso, catarata e hipoglicemia 31. La ictericia prolongada puede ser una manifestación de otras patologías metabólicas, que una vez se haya descartado la atresia biliar, deben tenerse en cuenta como diagnóstico diferencial incluyendo la enfermedad de Gaucher, el síndrome de Alagille o la enfermedad de Neimann Pick 32,33.
La pléyade de manifestaciones clínicas se extiende a sistemas endocrinológicos, cutáneos y aspectos hematológicos, de donde cabe destacar las anormalidades en el control glicémico y las citopenias, las cuales son claves en el diagnóstico de EIM, en conjunto con la determinación del balance acido básico, el estado de los electrolitos y determinación de la respuesta del paciente a su estado catabólico 34,35. Este conjunto de manifestaciones, cuando se circunscribe a la urgencia metabólica del neonato, pueden agruparse inicialmente en tres grandes grupos, a partir de las cuales se puede dirigir el trabajo diagnóstico. Esto grupos son la acidosis metabólica, la hiperamonemia y la hipoglicemia, las cuales se relacionan con la presentación de encefalopatía aguda de origen metabólico.
Acidosis metabólica
La acidosis metabólica es la consecuencia de un proceso fisiopatológico en el que se añade ácido o se elimina álcali de los líquidos corporales 36,37 y puede suceder como resultado de la acumulación endógena de ácidos que consumen el bicarbonato (acidosis metabólica con brecha aniónica aumentada) o por la pérdida de bicarbonato del tracto gastrointestinal o renal (hiperclorémica o acidosis metabólica de brecha aniónica normal) 38. El cálculo de la brecha aniónica es fundamental en el estudio de la acidosis y los EIM. El incremento en la brecha (>16) se observa en múltiples EIM; sin embargo, la acidosis con brecha aniónica normal, se limita a dos etiologías, la diarrea o la acidosis tubular renal (ver valores de referencia en la tabla 2) 39,40.
Tabla 2 Valores de referencia. Gases arteriales, amonio, glicemia central.
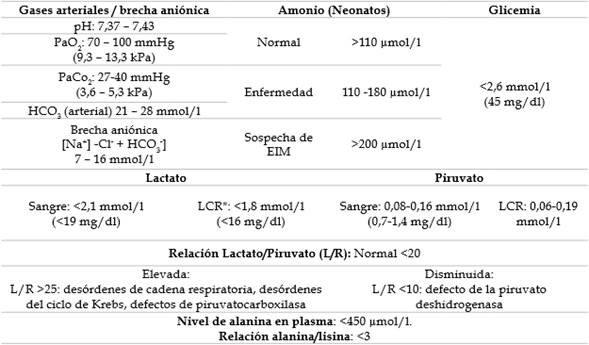
* LCR: líquido cefalorraquídeo.
El incremento de la brecha aniónica indica la presencia de una sustancia que ‘separa’ los cationes de los aniones, es decir, se incrementa la brecha o diferencia entre ambos grupos de electrolitos. Para el caso de los EIM de presentación aguda, asociados a la acidosis metabólica con brecha aniónica aumentada, se deben tener en cuenta principalmente, los valores de los cuerpos cetónicos (Acetoacetato, 3-hidroxibutarato y acetona) y el ácido láctico 40,41.
En los estados deficientes de glucosa, el metabolismo de los ácidos grasos estimula la acumulación del acetoacetato, el cual se reduce en la mitocondria produciendo 3-hidroxibutarato, ambas sustancias trasportan energía desde el hígado hacia los tejidos periféricos 42. La acetona se forma de la decarboxilación espontánea del acetoacetato. Los cuerpos cetónicos son una fuente alterna de energía para el SNC durante periodos prolongados de ayuno o desnutrición, su presencia indica por tanto que la β-oxidación responde ante la ausencia de fuentes de energía derivadas de los carbohidratos 43; sin embargo, si persiste la cetosis, una vez sea corregida la deficiencia de carbohidratos, esta se debe usualmente a desórdenes del catabolismo de los cetonas como la deficiencia de la succinil-CoA 3-cetoacido CoAtrasferasa y de la acetoacetil-CoAtiolasa 44.
Entre las acidemias orgánicas con brecha aniónica aumentada, se destacan las acidemias metilmalónica, propiónica e isovalérica 45,46, las cuales se relacionan con estado de hiperglicemia, ácido láctico normal o elevado e hiperamonemia (figura 1), lo que demuestra cómo deben correlacionarse los diversos estados de las vías metabólicas para orientar el diagnóstico 47.
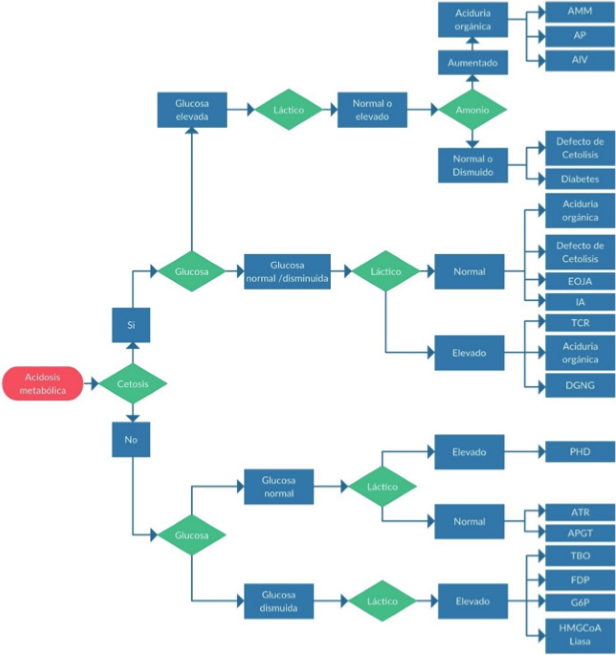
Figura 1 Flujograma diagnóstico a partir de la determinación clínica y paraclínica de la acidosis orgánica. AMM: acidemia metilmalónica. AP: acidemia propiónica. AIV: acidemia isovalérica. EOJA: enfermedad orina con olor a jarabe de arce. IA: insuficiencia adrenal TCR: Trastornos de cadena respiratoria. DGNG: Defectos de gluconeogénesis. PHD: déficit de piruvato deshidrogenasa ATR: acidemia tubular renal. APGT: acidemia piroglutámica TBO: trastornos de la β-oxidación. FDP: déficit de fructosa difosfatasa. G6P: déficit de glucosa 6-fosfatsa HNGCoA: deficiencia de hidroxi-metil-glutaril coenzima A liasa. En el caso de glucosa normal o disminuida con ácido láctico elevado, diversas acidemias orgánicas pueden explicar el cuadro.
Además de las alteraciones de los cuerpos cetónicos, el lactato en plasma está elevado en diversos EIM, que incluyen desde trastornos de la cadena respiratoria (enfermedades mitocondriales) hasta algunas acidemias orgánicas en donde se presenta interferencia con el metabolismo de la coenzima A (48, 49). El lactato elevado es una señal de hipoxia y compromiso del metabolismo energético y puede ser una causa de acidosis metabólica. La interpretación del lactato elevado debe ser cautelosa, dado que diversas maniobras, asociadas al manejo del neonato en estado agudo o complicaciones del estado encefalopático, pueden ser causa secundaria de su elevación, como por ejemplo el uso del torniquete para toma de muestra de sangre (tabla 3), ventilación asistida, actividad muscular (convulsiones), hipoxia central o periférica, falla renal, falla hepática, sepsis, cardiomiopatía, síndrome tubular renal, hipercloremia, infecciones urinarias y deficiencia de tiamina, entre otras (48, 50-53). La elevación del lactato tiene relevancia si se relaciona con el piruvato, dado que permite evaluar el estado de óxidoreducción; sin embargo, dados los falsos positivos por manipulación del tejido en la toma de muestra, es posible utilizar los niveles de alanina en plasma para evaluar directamente la concentración del piruvato (e indirectamente del lactato), dado que los niveles de alanina no se afectan por el torniquete (tabla 3). La figura 1 ofrece un flujograma que relaciona la acidosis con el estado de la glucosa, el ácido láctico y el amonio. Un mayor detalle frente al diagnóstico de hiperamonemia se aprecia en la figura 2.
Tabla 3 Ayudas diagnósticas para el manejo de los EIM. Se especifican recomendaciones para la toma y manejo de la muestra y el tiempo de respuesta máximo para casos de emergencia como es el caso de la UCIN. Se indica un tiempo de reporte estimado, varía de laboratorio en laboratorio.
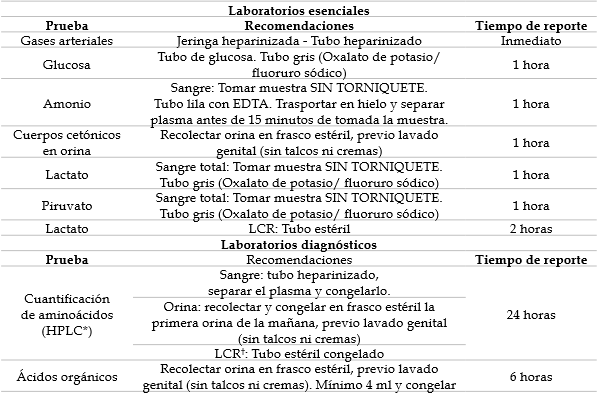
*Líquido cefalorraquídeo
† High Performance Liquid Chromatography.
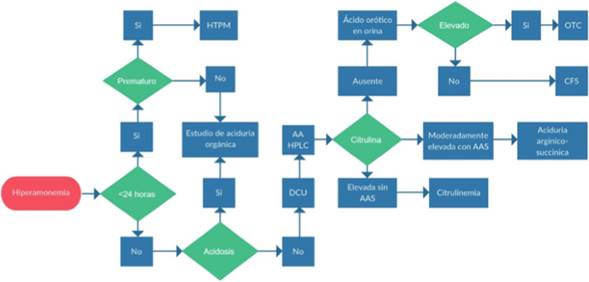
Figura 2 Flujograma diagnóstico a partir de la hiperamonemia. HTPM: Hiperamonemia transitoria de la prematurez. AA HPLC: Cuantificación de aminoácidos por HPLC (High Performance Liquid chromatography) DCU: defectos del ciclo de la Urea. AAS: ácido arginino-succínico. OTC: ornitina transcarbamiliasa. CFS: carbamoil fosfato sintetasa.
Hiperamonemia
La hiperamonemia es la elevación sintomática del amonio en sangre como resultado de diversas alteraciones bioquímicas, en algunos de los múltiples pasos del catabolismo de los aminoácidos, hacia su producto final, la urea (defectos del ciclo de la urea) 19. También puede deberse a inhibición secundaria del ciclo de la urea en ciertas acidemias orgánicas, desórdenes de la oxidación de ácidos grasos, deficiencia de piruvato carboxilasa y ciertos casos de hiperinsulinismo 54. Su reconocimiento y tratamiento, especialmente en el período neonatal es una emergencia clínica, dado que el efecto neurotóxico es directamente proporcional al tiempo de exposición tisular a niveles elevados de amonio 55, por lo que se debe proceder a su medición en todo neonato encefalopático 19.
Ante niveles elevados de amonio en el neonato (incluso mayores a 1.000 mmol/l), el diagnóstico más importante a tener en cuenta es un defecto del ciclo de la urea y como segunda opción alguna de las acidemias orgánicas, así como el diagnóstico diferencial de la HTPM (hiperamonemia transitoria del prematuro) 56, mientras que la presencia de hiperamonemia en lactantes mayores puede indicar desórdenes de la oxidación de ácidos grasos 57,58. El tiempo del inicio de los síntomas puede ser importante, los pacientes que presentan síntomas antes de las 24 horas de vida se relacionan con academia glutárica tipo II o deficiencia de piruvatocarboxilasa. Los síntomas antes de las 24 horas también son característicos de la HTPM, mientras que los síntomas después de las 24 se relacionan con defectos del ciclo de la urea u otras acidemias orgánicas 59. Sin embargo, se debe tener en cuenta que la dificultad de determinar el inicio de los síntomas y la dificultad de determinar con precisión el inicio del incremento de los niveles de amonio en las primeras horas de vida, obliga a que el trabajo diagnóstico tenga en cuenta los tres grupos diagnósticos en todo momento. De otra parte, se debe tener en cuenta que la acidosis metabólica no es un hallazgo típico de los defectos del ciclo de la urea, mientras que la cuantificación de aminoácidos en plasma es muy útil en clasificarlos 59,60. En la figura 2 se aprecia como la medición de ácido orótico y citrulina determinan el principal defecto enzimático.
Hipoglicemia
La hipoglicemia o disminución de los niveles plasmáticos de glucosa es el desorden metabólico más frecuente del neonato 61, se relaciona con EIM en casos de desorden de la oxidación de ácidos grasos, enfermedad de la orina con olor a jarabe de arce, galactosemia, acidemias orgánicas, desordenes de depósito de glucógeno y desórdenes de la gluconeogénesis 62. La sospecha clínica y la definición de acidosis metabólica, hiperamonemia e hipoglicemia, permite orientar la solicitud e interpretación de las ayudas diagnósticas, las cuales incluyen desde análisis de laboratorio de rutina hasta estudios de mayor complejidad.
Los desórdenes metabólicos más conocidos, con que se relaciona la hipoglicemia, son los desórdenes del metabolismo de carbohidratos y los desórdenes de la β-oxidación. La hipoglicemia en los desórdenes del metabolismo del glucógeno se debe principalmente a la incapacidad de hepática de liberar glucosa a partir del glucógeno, la cual se hace crítica en los periodos de ayuno 63. Tanto la hepatomegalia y la hipoglicemia así como la acidosis láctica, son características de este tipo de desórdenes de presentación aguda como la glucogenosis tipo I o enfermedad de Von Gierke (deficiencia de glucosa 6-fosfatasa); sin embargo, es de resaltar que la hipotonía, la cardiomegalia y la hepatomegalia sin hipoglicemia sugieren otro tipo de trastorno del metabolismo del glucógeno, tal como es glucogenosis tipo II o enfermedad de Pompe causada por deficiencia de la alfaglucosidasa ácida 63. No se deben descartar otros trastornos como la galactosemia y la intolerancia hereditaria la fructosa, aunque los síntomas de esta última solo se presentan luego de la introducción de fructosa en la dieta 64.
Ayudas diagnósticas
Los valores normales de las pruebas de laboratorio que definen la acidosis, la hiperamonemia y la hipoglicemia se aprecian en la tabla 2. Algunas características de las pruebas que contribuyen al definir el diagnóstico y evolución de un paciente con EIM, se describen en la tabla 3, incluyendo recomendaciones para la toma de muestra. Estos laboratorios esenciales permiten establecer el estado metabólico del paciente y orientan la solicitud e interpretación de estudios que definen la etiología del trastorno. En la misma tabla se describen estudios de orden diagnóstico; sin embargo, la lista no es exhaustiva dado que no se consideran algunos tests adicionales como las pruebas moleculares o las pruebas de actividad enzimática. Los tests esenciales están disponibles en la mayoría de hospitales que tienen la complejidad para albergar una UCIN y su interpretación orienta el siguiente nivel de estudio. En la figura 1 se aprecia al flujograma diagnóstico general a partir del diagnóstico de acidemias metabólicas. En la figura 2 se aprecia en detalle el flujograma de estudio de la hiperamonemia.
Manejo de muestras en casos de fallecimiento y sospecha de EIM
Es frecuente que el desenlace clínico de los pacientes sea letal, debido a la presentación aguda y severa de estas enfermedades, la cual no permite alcanzar en una gran proporción de los sujetos afectados, un diagnóstico definitivo 65,66; sin embargo, esa situación no impide que las investigaciones diagnósticas no puedan realizarse post mortem, lo cual es imperativo para alcanzar un diagnóstico que conlleve a una asesoría genética apropiada, a la planeación de un próximo embarazo y la atención de un neonato que no tenga la incertidumbre de la falta de diagnóstico, en un caso familiar previo 67. En caso de fallecimiento del paciente y ante la sospecha de EIM, se deben guardar muestras biológicas que pueden ser usadas para diversos análisis bioquímicos y genéticos 68. Las recomendaciones de toma de muestra se muestran en la tabla 4. Cabe recalcar que la toma de muestras debe hacer con previo consentimiento de los padres, explicando las razones para su necesidad y la importancia de hacer todo lo posible por un diagnóstico definitivo.

CONCLUSIONES
Los EIM en la UCIN deben hacer parte del trabajo diagnóstico de todo neonato con manifestaciones neurológicas o manifestaciones atípicas en otros sistemas, en quienes se hayan descartado noxas infecciosas o hipóxicas; sin embargo, la sospecha de EIM debe ser temprana, debido a que el retraso en su diagnóstico y su posterior manejo tiene consecuencias devastadoras para el paciente. Los paraclínicos para clasificar el tipo de trastorno se encuentran a disposición en la mayoría de centros en donde opera una UCIN, de tal modo que se deben interpretar en el contexto de la sospecha de EIM y teniendo en cuenta las precauciones para la toma de muestra. Así mismo, en caso de no alcanzar diagnóstico en vida, se deben tomar las medidas de toma de muestra luego del deceso.

