











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La secuenciación de nueva generación (Next Generation Sequencing [NGS]) es un grupo de tecnologías diseñadas para secuenciar gran cantidad de segmentos de ADN de forma masiva y en paralelo, en menor cantidad de tiempo y a un menor costo por base (1,2). Su uso se dio inicialmente para detectar variantes de nucleótido único y cada vez se ha desarrollado para otro tipo de variantes, como inserciones, deleciones y grandes rearreglos. Gracias a los recientes desarrollos en las pruebas basadas en NGS, estas tecnologías se plantean como estrategias de gran utilidad para la prevención, el diagnóstico, el tratamiento y el seguimiento de un amplio espectro de enfermedades, incluidas condiciones genéticas, patologías crónicas y enfermedades infecciosas, y se prevé que en un futuro cercano su creciente aplicación clínica generará resultados favorables para lograr el diagnóstico molecular en un número mayor de pacientes y a un menor costo (3-5). En la práctica clínica es creciente el uso actual de las pruebas basadas en NGS; sin embargo, aún existe incertidumbre sobre aspectos importantes como las indicaciones adecuadas para su uso, limitaciones de la técnica (sensibilidad y especificidad), reporte de variantes, interpretación de resultados, relación costo-beneficio y cobertura en el Plan Nacional de Salud (6).
En esta revisión queremos presentar de manera clara los conceptos básicos que definen las tecnologías NGS, las consideraciones de su uso actual en la práctica clínica y perspectivas a futuro.
Desarrollo
Pruebas NGS: desarrollo histórico y conceptos básicos
Desde 1977, la secuenciación Sanger ha sido la prueba estándar para la detección de variantes en el ADN. Su precisión y relativamente sencillo análisis de datos han permitido su uso en el diagnóstico de enfermedades monogénicas; sin embargo, en enfermedades que exhiben heterogeneidad genética, donde se requiere la secuenciación de múltiples genes, el proceso de secuenciación se hace largo y tedioso (4), lo que ha generado ansiedad en las familias y retraso en el tratamiento. Además, el precio para secuenciar todo el genoma humano era muy elevado, de aproximadamente 300 millones de dólares, lo que dificultaba su aplicación (7,8). Posteriormente, y debido a la necesidad de analizar fragmentos más grandes de ADN y agilizar el proceso de secuenciación, surgió la técnica de secuenciación en escopeta (shotgun sequencing), a partir de la cual fragmentos superpuestos de ADN se secuenciaban por separado y luego se ensamblaban en una sola secuencia continua (9). Este avance, y otros, fueron usados y permitieron la culminación del proyecto de genoma humano (9). Desde el 2005, se inició la comercialización de las pruebas NGS (10), las cuales han revolucionado la forma en la que se secuencian los genomas. Esta nueva tecnología ha permitido la lectura de millones de secuencias de forma masiva y paralela en un menor lapso y a un menor costo por base, facilitando el diagnóstico de enfermedades de alta heterogeneidad genética y su aplicación en la práctica clínica (11,12). Adicionalmente, permite, por ejemplo, secuenciar, un panel de genes específicos, la porción genética codificante completa (exoma) o todo el genoma completo de un individuo en 1 o 2 días con un costo aproximado de 5000 dólares (13).
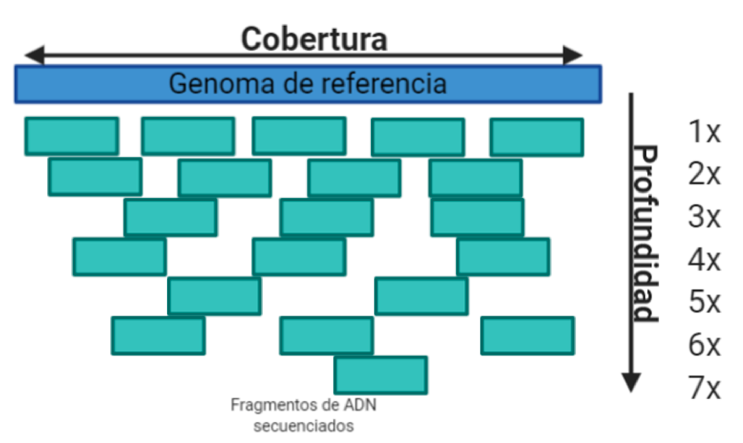
Hay dos conceptos que son fundamentales para entender el proceso y los resultados de las pruebas basadas en tecnologías NGS: cobertura y profundidad. Existe aún confusión en la utilización de estos términos, que comúnmente son usados de manera indistinta en la literatura. La cobertura (coverage o breadth of coverage, en inglés) se refiere al porcentaje de bases del genoma de referencia que están siendo secuenciadas una cantidad determinada de veces (14). Por otro lado, la profundidad (depth o depth of coverage) representa el número promedio de veces que cada base en el genoma es secuenciada en los fragmentos de ADN (14) (figura 1). Los valores apropiados de profundidad dependerán de la aplicación de la técnica de secuenciación (panel de genes, exoma o genoma) y la frecuencia alélica de las variantes que se analizarán (germinales y somáticas).
Técnicas actuales de mayor uso de secuenciación de nueva generación
Todas las técnicas NGS comparten la capacidad de secuenciar una gran cantidad de fragmentos de ADN de forma paralela en un corto lapso. Para lograr este objetivo, siguen un abordaje metodológico semejante que se puede resumir en cinco pasos: 1) segmentación del ADN en varios fragmentos, 2) marcaje del ADN por medio de primers o adaptadores que indican el punto de partida para la replicación, 3) amplificación de los fragmentos de ADN marcados con adaptadores por métodos basados en reacción en cadena de la polimerasa (PCR, por sus siglas en inglés), 4) secuenciación o lectura de los fragmentos de ADN y 5) reconstrucción de la secuencia completa por medio de secuencias de referencia y exportación a ficheros de almacenamiento de datos (4,15). En los siguientes apartados se revisarán los métodos de secuenciación Illumina e Ion Torrent, que son las dos tecnologías de mayor utilización actualmente (tabla 1).

Secuenciador Illumina
La secuenciación por medio de Illumina se caracteriza básicamente por la ejecución de los siguientes procesos:
La amplificación de los fragmentos de ADN para la generación del clúster (colonias del mismo fragmento) se realiza mediante el método de PCR en puente.
La detección de bases en la secuenciación se hace a través de etiquetas fluorescentes.
Generación del clúster
Este proceso se logra mediante el método de amplificación en puente (figura 2). Los fragmentos de ADN se colocan sobre una superficie sólida de vidrio separada por carriles. Cada carril está completamente recubierto por oligonucleótidos complementarios a los adaptadores de cada fragmento que se va a secuenciar, por lo que permiten que cada fragmento se pueda anclar a la celda de flujo.

Elaboración propia
Figura 2. Generación de hebras mediante PCR en puente.a) El fragmento de ADN se ancla a la celda de flujo por medio de la unión de sus adaptadores a los oligonucleótidos complementarios. b) La polimerasa genera una hebra reversa complementaria y la hebra original es retirada. c) La hebra reversa se pliega y queda en forma de puente, donde la polimerasa genera una hebra complementaria idéntica a la original. d) El proceso se repite masivamente.
Una vez anclados los segmentos, la polimerasa inicia el proceso de copia en la hebra de ADN y genera una hebra reversa complementaria. La hebra original es entonces retirada; mientras que la hebra reversa, a través de una secuencia terminal, se pliega y se ancla a su respectiva secuencia complementaria de oligonucleótido, y así queda en forma de puente. Posteriormente, la polimerasa genera una hebra complementaria idéntica a la original, que resulta en dos hebras clonadas del segmento inicial. Este proceso se repite masivamente hasta formar millones de copias de cada fragmento (16-18).
Secuenciación
Al finalizar la amplificación clonal, se retiran todas las hebras reversas y quedan únicamente las hebras idénticas a las originales. En este punto, en la placa se introducen nucleótidos modificados con etiquetas fluorescentes específicas para cada tipo. Los nucleótidos utilizados presentan una modificación química (terminadores reversibles) que evita la unión de más de un nucleótido marcado en cada sitio de reacción, de tal manera que se puede ubicar el que corresponde a cada punto en la secuencia y se disminuye el riesgo de errores en la secuenciación. Cada vez que una base se adhiere emite una fluorescencia propia que permite su identificación. La etiqueta debe ser removida antes de la colocación del siguiente nucleótido para evitar que dos bases emitan señal a la vez. Al finalizar la primera lectura, el fragmento resultante es retirado. Este paso se repite simultáneamente con todas las hebras del mismo clúster de forma paralela hasta completar la secuenciación (16-18).
La exactitud de la secuenciación en el secuenciador Illumina será determinada por la intensidad de la señal, y la longitud de las lecturas, por el número de ciclos realizados. Actualmente, alcanzan una longitud por lectura de hasta 300 pares de bases (3).
Secuenciador Ion Torrent de Thermo Fisher
La secuenciación en Ion Torrent se lleva a cabo en un chip semiconductor, y se diferencia de Illumina en los siguientes aspectos:
Reacción en cadena de la polimerasa en emulsión
Cada fragmento de ADN se deposita en micromicelas ubicadas en una emulsión de aceite en agua, donde, además, se deposita una microesfera que está recubierta de adaptadores complementarios a los adaptadores del fragmento, y una polimerasa que iniciará la PCR (figura 3). Para ser copiada la hebra original se hibrida con los adaptadores ubicados en la microesfera. Después de la amplificación, se retiran todas las hebras complementarias y se dejan solo las hebras con los adaptadores ligados a la microesfera, y finalmente quedan miles de copias de un mismo fragmento de ADN (17). Este paso se repite en millones de esferas que posteriormente se colocan cada una por separado en micropocillos que contienen todos los componentes necesarios para iniciar la secuenciación (19).

Secuenciación mediante ion semiconductor
Cuando un nucleótido es ligado a la hebra que se va a secuenciar, se forma un enlace covalente y se libera un ion de hidrógeno cargado positivamente. Cada vez que se libera un ion de hidrógeno, se da un cambio en el pH de la solución del pocillo y se genera una corriente eléctrica. Este método se basa en la detección del cambio de voltaje por medio un sensor llamado ISFET (Ion-Sensitive Field-Effect Transistor), que indica que el nucleótido se ha incorporado correctamente (20). Para la identificación del tipo de nucleótido agregado, inicialmente se inserta solo un tipo de base en el pocillo y el sensor ISFET detectará o no el voltaje dependiendo de la complementariedad de la base. Si esta no es complementaria, se agrega otro nucleótido hasta que se detecte voltaje. En esta técnica, los nucleótidos no están modificados con terminadores reversibles, por lo que en caso de que haya homopolímeros (dos o más nucleótidos iguales que se repiten), el pH disminuirá en mayor proporción y la diferencia de voltaje aumentará proporcionalmente al número de bases añadidas (19). Este proceso ocurre de forma paralela en todos los pocillos del chip y alcanza una longitud de lectura de hasta 400 pares de bases (17,19).
Aplicación de las tecnologías basadas en secuenciación de nueva generación en la práctica clínica
Secuenciación de panel de genes
Es una prueba que secuencia un número determinado y específico de genes que están relacionados con una enfermedad o grupo de enfermedades. Supone un ejercicio clínico previo que esté enfocado en un grupo de patologías similares y que se beneficie de esta herramienta para el diagnóstico diferencial. Se recomienda su uso cuando en la clínica del paciente se observe un fenotipo característico y se sospeche de una enfermedad genética que sea causada por una lista determinada de genes candidatos con buena posibilidad para establecer el diagnóstico molecular (véase el caso clínico). Es la prueba de menor costo, se realiza más rápidamente, tiene una profundidad de secuenciación mayor y la interpretación y análisis de resultados es más sencilla, debido a la existencia de bases de datos de referencia que asocian las variantes halladas en los genes con la enfermedad relacionada (15,21). Una desventaja es que la información genética de la población latinoamericana está subrepresentada tanto en las bases de datos poblacionales como en las específicas de enfermedad, por lo que se dificulta la interpretación clínica de las variantes encontradas en pacientes con esta ancestría (22). Existen bases de datos como el Genetic Testing Registry (23), que reúne la información de los paneles genéticos específicos y validados con la información suministrada por los laboratorios proveedores. Se han implementado en la práctica clínica paneles NGS para diferentes condiciones; tal es el caso del panel Hereditary Cancer SolutionTM (24), que evalúa 26 genes asociados con cáncer de seno y ovario, cáncer colorrectal hereditario no polipósico y síndrome de poliposis intestinal.
Secuenciación completa de exoma
En esta prueba se secuencian todas las regiones codificantes del genoma, es decir, el exoma, el cual representa entre el 1 y el 2 % de la secuencia genómica completa y donde se hallan hasta el 85 % de las variantes genéticas reportadas como patogénicas (25). Es de gran utilidad en pacientes que padecen enfermedades con gran heterogeneidad genética, como el autismo, o que presenten un fenotipo complejo, donde el cuadro clínico no es lo suficientemente característico de una variante o de uno o varios genes específicos (26). Comparada con la secuenciación completa del genoma, es menos costosa, su duración es menor y la interpretación y análisis de datos es menos compleja. Presenta una amplia capacidad para la identificación de enfermedades genéticas; sin embargo, se debe tener presente que no está diseñada para identificar variantes patogénicas que se encuentren en regiones no codificantes (regiones de ADN que no tienen información que codifica para la proteína) como intrones o regiones promotoras (21,27). La interpretación de los resultados puede ser más compleja y puede ser más probable encontrar variantes de significado incierto o variantes que no se correlacionen con la clínica del paciente, en cuyo caso el estudio de familiares —como el exoma trío, es decir, secuenciación de exoma del probando y sus dos progenitores— podría resultar útil (15). Su uso clínico se ha propuesto, por ejemplo, como primera prueba diagnóstica en trastornos del neurodesarrollo (28).
Secuenciación completa de genoma
Esta prueba evalúa todas las bases del genoma, incluyendo las regiones no codificantes. Tiene como ventaja que es la mejor prueba para la detección de nuevas variantes genéticas y estructurales que no se asociaban previamente con el cuadro clínico que se iba a estudiar (15). Es de gran utilidad en enfermedades genéticas raras en las que no es posible llegar al diagnóstico por medios convencionales, es decir, en las que hay odisea diagnóstica y cuando el fenotipo del paciente pueda ser explicado por una variante de novo. Además, es la prueba con menos sesgos durante el análisis de genes, debido a que la secuenciación no está dirigida hacia genes específicos (3,16,29). En la actualidad, su uso sigue siendo poco frecuente, debido a su mayor costo, mayor tiempo de entrega de informe, falta de anotaciones sobre regiones no codificantes y la dificultad en el análisis e interpretación de sus resultados (30). Está en desarrollo su implementación en la práctica clínica, especialmente en población pediátrica, aunque existe aún debate sobre la utilidad clínica y la costoefectividad de este tipo de pruebas (31).
Aspectos éticos y normatividad sobre el reporte, interpretación y validación de hallazgos
Para la correcta interpretación de datos, en primer lugar, se debe verificar la calidad de los datos secuenciados, mediante el uso de un genoma de referencia validado y conocido. Cuando este se alinea con la muestra secuenciada, permite identificar errores cometidos durante la secuenciación y el análisis de los datos (32). Posteriormente, se deben identificar las variantes genéticas y definir si estas se asocian con alguna enfermedad.
El Colegio Americano de Genética y Genómica Médica recomienda el uso de la siguiente terminología para el reporte de datos de las variantes genéticas identificadas: patogénica: variantes que tienen evidencia fuerte de asociación con enfermedad. Probablemente patogénica: variantes que probablemente están implicadas con enfermedad, pero no hay evidencia suficiente para demostrar asociación. De significado incierto (VUS): variantes con posibles cambios funcionales, pero con evidencia contradictoria o insuficiente como para considerarla benigna o patogénica. Probablemente benigna: variantes con evidencia que sugiere benignidad, pero con datos débiles en la literatura que no descartan impacto biológico y eventualmente clínico. Benigna: variantes genéticas que no alteran funcionalidad (21,33).
La interpretación de cada resultado en las pruebas NGS es compleja, ya que, al secuenciar una mayor cantidad de genes, se pueden evidenciar un mayor número de variantes genéticas incidentales que no presentan significado conocido para la definición de un plan de acción (34). La mayoría de estas variantes desconocidas pueden no conceder riesgo en un individuo sano, pero también existe la posibilidad de que sea una nueva variante patogénica (35). Se ha reportado que, en un individuo sano, del 100 % de variantes encontradas, solo el 60 % tiene probabilidades reales de asociarse a enfermedad (36). Las VUS, aunque actualmente no pueden ser clasificadas como causales de enfermedad, pueden cambiar su clasificación según la aparición de nueva evidencia que las clasifique como patogénicas o benignas.
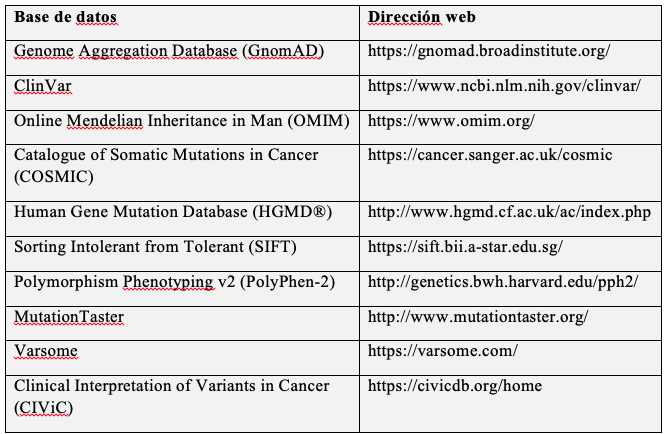
Los laboratorios deben seguir un plan claro y adecuado para la clasificación de significado clínico de las variantes, que incluya el uso de programas predictivos computacionales, búsqueda de variantes en bases de datos, literatura de casos clínicos relacionados con las variantes y evidencia de su efecto biológico en estudios funcionales. Los programas de bioinformática (por ejemplo, SIFT, PolyPhen o MutationTaster) utilizan algoritmos especiales que intentan predecir el efecto de la variante en la proteína y su posible impacto en la función (37,38). Los programas también evalúan si la variante interfiere o no en el proceso de splicing (corte de intrones y empalme de exones). El porcentaje de predicción de estos programas es de aproximadamente del 65 % al 80 % para variantes localizadas en regiones codificantes, lo que no garantiza que la predicción sea un 100 % correcta (39). En consecuencia, no es recomendable usar un solo algoritmo y se debe considerar evidencia adicional para realizar una afirmación clínica. Se debe identificar la variante encontrada en bases de datos y examinar su frecuencia y si presenta evidencia de estar asociada o no a patogenicidad. GnomAD, ClinVar, OMIM, COSMIC y Human Gene Mutation Database son bases de datos confiables para examinar la asociación de una variante a enfermedad (tabla 2) (33). Este proceso de interpretación deberá ser realizado idealmente por el laboratorio como parte de su servicio y explicado al paciente por profesionales con entrenamiento calificado en asesoría genética (40). En caso de que todavía existan dudas sobre la significancia clínica de los hallazgos o aún no se hayan identificado variantes que expliquen la clínica del paciente, se puede considerar como opción la secuenciación de los familiares del paciente para ayudar a clarificar la relación de la presencia de una variante con la presentación de la enfermedad (41). Además, se debe contemplar la recomendación de no entregar resultados definitivos basados en una solo prueba NGS, para disminuir la probabilidad de error, y en este sentido tener presente la posibilidad de confirmación de hallazgos con secuenciación Sanger u otras técnicas ortogonales, según el hallazgo (42,43).
Recientemente, la Food and Drug Administration (FDA) publicó su guía para el diseño, el desarrollo y la validación analítica de las pruebas basadas en NGS, diseñadas para el diagnóstico de enfermedades de línea germinal (44). Para la entrega de resultados, la FDA recomienda que las interpretaciones se seleccionen de bases de datos que estén correctamente validadas, curadas y actualizadas (43). Estas bases de datos deben posibilitar el acceso al público, con el fin de que tanto pacientes como profesionales de la salud revisen y comparen los datos. Todos los resultados deben cumplir con regulaciones federales, donde se certifique la seguridad de los datos y la privacidad del paciente. Adicionalmente, es mandatorio emplear una nomenclatura que se encuentre estandarizada para el reporte de resultados, con el propósito de facilitar su comparación con diferentes fuentes (43).
Es de suma importancia que sea confiable y claro el informe que reciba el paciente, junto con una explicación por parte del profesional a cargo sobre la implicación de este resultado, y con la posibilidad de brindar educación sobre la enfermedad y apoyo emocional, si es necesario (45). Es precisa una asesoría pretest antes de ordenar la prueba, con el fin de explicar al paciente en qué consiste y aclarar los alcances e implicaciones de su ejecución. Además, en la consulta inicial se deben obtener datos como: antecedentes personales y familiares, riesgos psicosociales relevantes, nivel de educación, conocimiento sobre la enfermedad, y llevar a cabo un proceso de consentimiento informado previo a la prueba (46). Al obtener los resultados, se deberá realizar asesoría postest, en la cual se clarifique el reporte obtenido a través de una atención personalizada que se adecúe a la condición de cada paciente y se exponga un claro significado clínico, enfocado en las posibles decisiones terapéuticas, indicando beneficios y complicaciones, y se solucionen dudas (figura 4). Las consultas generalmente tienen una duración de 30 a 60 minutos, aunque podrían llegar a necesitarse hasta 90 minutos para cubrir una atención apropiada (47-49). Esto dependerá del contexto individual de cada paciente.

Elaboración propia
Figura 4. Pasos para el correcto análisis, interpretación y entrega de resultados de NGSMLPA: amplificación de sondas dependiente de ligandos múltiples.
MLPA: amplificación de sondas dependiente de ligandos múltiples. Fuente: elaboración propia.
En el cuadro 1 se muestra un caso clínico real en el que se describe el manejo adecuado de un paciente que se somete a una prueba de NGS.
Cuadro 1. Caso clínico
Una paciente de 63 años de edad asistió a consulta de genética médica, remitida por el servicio de Oncología con el resultado de un panel NGS. Tiene historia personal de cáncer de ovario diagnosticado a los 33 años, manejado con cirugía y quimioterapia. A los 62 años, le fue diagnosticado cáncer de mama, con reporte patológico de carcinoma ductal in situ de alto grado de patrón comedo con microcalcificaciones sin evidencia de microinfiltración. Recibió manejo con cuadrantectomía más radioterapia adyuvante. En el momento de la consulta recibía tratamiento con tamoxifeno. También tenía una historia familiar recurrente de cáncer (madre, hermana, sobrina y tía materna con historia de cáncer de mama).
Se realizó un estudio mediante panel NGS de 28 genes relacionados con síndromes de cáncer hereditario. El reporte evidencia la presencia de la variante c.1188del (p.Val397Phefs*17) heterocigota en el gen CHEK2, reportada como patogénica para síndrome de cáncer de mama y ovario hereditario. Adicionalmente, se encontraron las variantes c.511A>G (p.Ile171Val) en el gen NBN y c.1564C>T (p.Pro522Ser) en el gen PALB2, ambas clasificadas como VUS.
El resultado de este reporte debe entregarse a la paciente, explicándole las implicaciones que tiene y los cambios que se pueden realizar en la prevención y el tratamiento, siempre brindando apoyo emocional. Se debe explicar que, aun cuando esta mutación del gen CHEK2 no tiene implicación farmacogenómica actual, sí es útil para establecer el plan de vigilancia y seguimiento de la enfermedad y permite realizar consejería genética a la familia. La paciente tiene riesgo de un segundo cáncer de mama dentro de los primeros diez años del primero de hasta un 29 % y un riesgo posiblemente elevado para cáncer colorrectal. Este resultado también representa una probabilidad del 50 % para cada uno de los hijos de la paciente de tener la misma mutación, lo que se podría traducir en un riesgo mayor de tener cáncer de mama para sus descendientes. Hay que tener en cuenta que el manejo en estos casos no debe estar basado en una sola prueba, sino que además se debe tener en cuenta la historia personal y familiar de cada paciente.
Las VUS, aunque actualmente no se pueden clasificar como causales con la evidencia disponible, pueden cambiar su clasificación según la aparición de nueva evidencia que las clasifique como patogénicas o benignas, por lo que es importante establecer un plan de reanálisis de datos con cierta periodicidad.
Fuente: elaboración propia.
Limitaciones
Las pruebas basadas en tecnologías NGS, como cualquier prueba de laboratorio, tienen limitaciones que deben ser conocidas para una adecuada interpretación de los resultados. El costo de una secuenciación veloz, masiva y paralela es el aumento de errores en relación con la cobertura o profundidad con la que se secuencia el gen. En las pruebas NGS, la cobertura de lecturas de un mismo fragmento es más baja en comparación con Sanger, por lo que hay más probabilidad de que se omitan regiones con variantes considerables para proporcionar un diagnóstico adecuado. Por otra parte, en el proceso de amplificación basado en PCR también se pueden generar errores, especialmente en regiones con alto contenido de guanina y citosina, que finalmente pueden terminan por afectar el posterior análisis de datos (50). Otra limitación es la reducción de la longitud de las secuencias (de 800-1000 pb en Sanger a 40-400 pb en NGS), lo que genera mayor dificultad en el análisis de datos (48). Dependiendo del diseño de la prueba, las pruebas basadas en NGS pueden presentar limitaciones en la identificación de deleciones, inserciones, mosaicismos y traslocaciones de gran tamaño o en la detección de variantes de número de copias, en los que serían de mayor utilidad otras pruebas más específicas como la amplificación de sondas dependientes de ligandos múltiples, la hibridación fluorescente in situ o la hibridación genómica comparativa (21). Adicionalmente, en los últimos años, con el uso de estas tecnologías, es cada vez mayor el reporte de variantes genéticas de significado clínico incierto, lo que suscita incertidumbre al analizar la información para determinar un diagnóstico y tratamiento apropiados (51,52). Estos errores se pueden disminuir implementando buenas prácticas de laboratorio que garanticen técnicas de buena calidad y a partir de la utilización de estándares específicos con evidencia científica válida en todas las bases de datos.
En Colombia, se espera que a futuro se realicen más estudios epidemiológicos que permitan identificar la asociación entre variantes genéticas propias y la enfermedad que se va a estudiar, y que se generen datos genómicos de consulta abierta. Actualmente, en Colombia solo se dispone de información genómica de nuestra población en una base de datos pública, perteneciente al proyecto 1000 genomas, que contiene variantes alélicas identificadas a partir de 148 muestras individuales de ADN, recolectadas en la ciudad de Medellín (53). Todas las pruebas aquí nombradas están cubiertas por el Plan de Beneficios en Salud (PBS) y se pueden solicitar sin costos adicionales para el paciente. De acuerdo con la última Clasificación Única de Procedimientos en Salud, Resolución 5851 del 21 de diciembre del 2018. Las pruebas disponibles para genética se pueden ubicar desde el código 90.8.4 hasta el código 90.8.4.41 (54).
Perspectivas
La secuenciación de nueva generación es una herramienta que ya se está utilizando y que apoya el diagnóstico de pacientes con enfermedades comunes como el cáncer hasta pacientes con enfermedades raras o huérfanas. En la práctica clínica, su implementación genera retos que deben considerarse para que el impacto positivo de estas tecnologías sea aún mayor. Estos incluyen reducir el tiempo de entrega de resultados, generar guías para el manejo clínico de las variantes, aumentar la representación poblacional en las bases de datos y estandarizar las prácticas de laboratorio y de análisis de datos (55). Asimismo, es importante que exista legislación sobre el uso de las tecnologías de secuenciación en el ámbito clínico, con el fin de que se estandaricen los procesos y se promueva la implementación e investigación sobre el tema. Los gobiernos de algunos países, como China y Estados Unidos, han adelantado medidas regulatorias y guías con esta finalidad (56).
La investigación en tecnologías de secuenciación de ADN se encuentra en continuo desarrollo. Actualmente, se enfoca en mejorar la interpretación de los datos y la clasificación de las variantes, para lo cual existen, por ejemplo, iniciativas dirigidas al uso de la inteligencia artificial en la detección de variantes y la predicción de su efecto (55). Han surgido también tecnologías llamadas secuenciación de tercera generación, como la secuenciación por Nanopore, que se caracteriza por la generación de fragmentos de lectura largos (de hasta 1 Mb) sin necesidad de amplificación por PCR, lo que evita sesgos y permite una cobertura mucho más homogénea del genoma (57). Estos avances se verán implementados en la práctica clínica en un futuro no muy lejano.
Conclusiones
Las NGS son pruebas paraclínicas que siempre deben partir de una aproximación clínica inicial adecuada y de una visión integral del paciente que incluye resultados de otras pruebas paraclínicas para su uso razonado, correcta interpretación y toma de decisiones acertadas en la práctica médica. En la última década, las pruebas NGS han establecido su valor como prueba diagnóstica, dado su buen rendimiento para la detección de enfermedades genéticas y el descubrimiento de nuevas variantes patogénicas. Esta tecnología ha permitido facilitar el diagnóstico molecular, al ser una herramienta más eficiente y veloz en la secuenciación de genes, y ha facilitado la identificación y la clasificación de múltiples variantes genéticas junto con su respectiva asociación patológica. Adicionalmente, se han disminuido los costos de la secuenciación y la duración en la que se define el diagnóstico, lo que permite establecer oportunamente medidas de prevención en pacientes con alto riesgo a futuro.
Las pruebas basadas en NGS representan retos y desafíos, como la comprensión de su utilización de forma adecuada, su validación a partir de estándares de calidad y la interpretación correcta de variantes reportadas, junto con una asesoría acertada para el paciente, con el fin de lograr que el diagnóstico de enfermedades de base genética sea cada vez más oportuno y adecuado. Es de gran trascendencia que los médicos tengan la información básica para poder solicitar e interpretar estas pruebas, dada su relevancia clínica actual.

