











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
In 1875, M. Raymond described the onset of progressive muscle atrophy and weakness in a series of survivors of acute poliomyelitis during childhood. At that time, Jean-Martin Charcot stated that the initial infection may make the neurons of these individuals more sensitive, so that spinal diseases occurred subsequently, resulting in a new weakness as a consequence of the overuse of the affected muscles. In 1979, after the publication of a report made by an elderly patient about the motor difficulties he developed after suffering from polio in his childhood, a significant increase of the amount of individuals with similar clinical pictures was observed. The term post-polio syndrome (PPS) was finally established around the 1980's. 1-3
Although this is a little known entity, PPS can affect between 20% and 85% of the population with new neuromuscular symptoms that occur at least 15 years after achieving stability in patients with previous acute paralytic poliomyelitis. 4 In 2000, the first diagnostic criteria were described 3,5, which included 1) suffering from paralytic poliomyelitis with evidence of motor neuron loss, confirmed by clinical history, and symptoms and signs of chronic denervation in electromyographic study (EMG); 2) partial or complete recovery of the acute disease, accompanied by a period of about 15 years of stable neurological function; 3) sudden or gradual onset of muscle weakness or abnormal, persistent and progressive fatigue, atrophy or muscle or joint pain; and 4) persistent symptoms for at least a year.
Other neurological, medical or traumatic causes that explain the clinical picture should be excluded, and some exclusion criteria should be considered, such as symptoms secondary to oropharyngeal or respiratory complications, structural radiculopathies, compressive neuropathies, degenerative arthropathies, joint instability, scoliosis and psychopathological symptoms such as anxiety syndrome, depression and sleep disorders. 6,7
Likewise, exploration should consider the presence of flaccid and asymmetric weakness with hyporeflexia in the affected areas, without associated sensory alterations. As part of the complementary studies, it is worth noting that there is no biochemical or physiological marker that allows for the unambiguous characterization of patients with PPS, so they are used mainly to identify or rule out other diseases. 1,8 Neither the analysis of the cerebrospinal fluid (CSF) nor blood analyses show alterations, although an increase in serum creatine kinase levels may be observed in a small number of patients, which is explained by muscle overstress. EMG shows signs of long-standing neurogenic involvement with loss of motor units that show an increase in polyphasia and amplitude, with giant potentials; even though this study identifies alterations compatible with previous poliomyelitis, it does not differentiate patients who report new symptoms. 9 In short, these studies are not useful to diagnose PPS, but to confirm that there are sequelae of polio and to rule out other pathologies such as neuropathies, radiculopathies and myopathies. 10-12
On the other hand, it is also possible to request a biopsy of the affected muscles, which can histologically show regrouping by types of fibers, atrophic angulated fibers and fibers with hypertrophy, which are characteristic of possible denervation or reinnervation. However, it is not usually useful for diagnosis. 13
Scientists from the National Institute of Neurological Disorders and Stroke (NINDS) and other institutions have shown, through several studies, that the weakness caused by PPS is a slow-evolving condition characterized by periods of stability, accompanied by a sudden deterioration in the ability to carry out daily activities. 14
The purpose of exposing the following cases is to present different associated diagnoses, clinical and diagnostic evidence, and an interdisciplinary management approach to this pathological entity.
CASE PRESENTATION
Case 1
59-year-old female patient, mixed race, from Popayán (Colombia), middle class, with a history of idiopathic scoliosis and polio during childhood, lumbar arthrodesis 26 years before consultation and dysplasia of the right hip. Pharmacological management of the patient included pregabalin and tramadol for chronic pain for 2 years. A recent clinical picture of 15 days of intense pain in the lumbar region that limits sitting and supine position and radiates to the lower left limb was reported, which is atrophic as a sequel to the childhood syndrome (Figure 1), with presence of lower extremity strength of 3/5 and positive reflexes. On admission to the emergency service, a foreign body sensation was reported in the right dorsal region, possibly associated with the history of arthrodesis.

Spine radiography was requested, revealing Harrington rod instrumentation without evidence of corrosion, left convex scoliosis in the lumbar spine, and wires and sublaminar hooks placed from T12 to S1. In addition, an electromyogram was indicated with nerve conduction velocities that show a chronic axonal injury in the lower limbs (Figure 2). The patient did not show signs of acute denervation or sensitive involvement, although muscle and joint pains persisted. The findings allowed ruling out radiculopathy and compressive neuropathy.

Figure 2 shows normal motor conduction velocities and amplitudes of motor evoked potentials, although the amplitude was lower in the left peroneum than in the right side by 50%, in spite of having normal parameters. The muscles of the upper limbs and the distal and dorsal latencies were normal; the amplitude and sensory conduction velocities of sensory potentials were also normal. The EMG showed signs of bilateral chronic denervation in the lower limbs, with a decrease in the recruitment of potential motor units and some polyphasic potentials.
After conducting clinical and paraclinical studies, it was determined that the symptomatology was associated with PPS since the necessary clinical diagnostic criteria were observed, as well as signs of chronic denervation in the lower limbs as reported by the EMG bilaterally, with a decrease in motor unit recruitment and some polyphasic potentials.
Therapeutic management with prednisone was initiated and a slight improvement of the symptoms was reported. Follow-up was performed for the following 2 months.
After conducting clinical and paraclinical studies, it was determined that the symptomatology was associated with PPS since the necessary clinical diagnostic criteria were observed, as well as signs of chronic denervation in the lower limbs as reported by the EMG bilaterally, with a decrease in motor unit recruitment and some polyphasic potentials. Therapeutic management with prednisone was initiated and a slight improvement of the symptoms was reported. Follow-up was performed for the following 2 months.
Case 2
47-year-old male patient, mestizo, from Popayán (Colombia), middle class, with a history of childhood poliomyelitis at age five, who consulted the emergency service due to sensation of hyperthermia, dyspnea and oppressive chest pain. Additionally, he presented cough with purulent expectoration, for which a chest X-ray was taken, finding consolidation in the left lung base. Possible left basal pneumonia was considered and management with ampicillin/sulbactam was initiated.
In addition to the initial symptoms, the patient reported fatigue when walking and myalgias with weakness in the left lower limb. Upon exploration of lower limbs, evidence of negative Patrick's test, intense muscular atrophy in the gastrocnemius and tibialis anterior muscles (with normal knee joint balance), lower extremity strength of 4/5, positive reflexes and preserved sensitivity were observed (Figure 3). Clinically, possible hip and sacroiliac joint pathology was ruled out. Although EMG was not performed, clinical differential diagnoses, such as a lumbar hernia, were ruled out through spinal magnetic resonance, which in turn ruled out nerve root compression.

Interconsultation with neurology was indicated, determining that the symptoms presented by the patient were associated with PPS after conducting clinical and paraclinical analyzes. Regardless of the lack of an EMG study, the diagnosis was achieved based on clinical findings, which are mandatory for this entity. Simultaneous symptomatic management with NSAIDs and therapies with physiotherapist were formulated, and the patient was monitored for the following 2 months. Adequate pain modulation and clinical evolution were evident at follow-up.
DISCUSSION
It should be noted that both patients gave their consent for conducting this academic and clinical study. It is possible to observe that the chronic pain presented by the patients had an adequate and satisfactory evolution, with clear and objective evidence of asymmetric muscle group atrophy with respect to the contralateral side. The current components of this clinical picture include progressive muscle weakness on the side of atrophy and generalized fatigue; these manifestations are compatible with PPS, which occurred approximately 40 years after the onset of the disease in both patients. 15-18
PPS should not be considered a sequel to polio. A sequence of a mutated genome in the poliomyelitis virus may be observed in the CSF of some patients; consequently, the presence of a chronic infection in infected neurons that prevails over the years is considered a mechanism. Although they are not the same entity, the most common complains about this syndrome include myalgias or muscle pains and arthralgias, as well as fatigue and low resistance to exertion. Diagnosis is difficult to achieve since the symptoms can be misinterpreted as related to the normal aging process. 19
Individuals with a history of poliomyelitis and suspected PPS should be referred to a specialist in neuromuscular disorders, preferably with experience in the treatment of people with post-polio. These specialists include, among others, neurologists, psychiatrists (rehabilitation specialists) and orthopedists. For a definitive diagnosis, different pathological entities should be ruled out, such as Parkinson's disease, cerebrovascular disease, multiple sclerosis, amniotrophic lateral sclerosis, radiculopathy, spinal cord tumors, comprehensive neuropathy, myasthenia gravis, muscular dystrophy, post-surgical processes, heavy metal poisoning, depression, among others. Likewise, a complete physical examination, such as that performed on the patients reported here, should be done -although only positive aspects were described-, as well as complementary tests. Although all the necessary tests were not carried out in these subjects, great value was given to data collection and clinical analysis, which allowed discarding a certain number of additional possibilities.
Differential diagnoses allow, in a certain way, to provide an accurate diagnosis at the moment of categorizing a pathology. For PPS, different tests should be considered, such as antinuclear antibodies to rule out an ongoing autoimmune process; cytochemical CSF study and viral panel to rule out herpetic encephalitis; electrophysiological study of the peripheral nerve; electromyography to rule out acute denervation processes such as radiculopathies or pathologies such as amyotrophic lateral sclerosis; magnetic resonance of the spine to rule out compressive lumbar hernias; simple computerized axial tomography of the skull to rule out ischemic strokes; and toxicological tests, since it is worth discarding heavy metal poisoning depending on the approach.
In short, the most useful way to approach this pathology is by building a good clinical history, including physical examination based on interdisciplinary approaches that allow understanding the patient at the time of requesting paraclinical test to discard any condition.
Most researchers agree that when the infectious event occurs, the motor neurons initiate cell death and, as a consequence, loss of innervation and motor function of the muscle fibers occurs, thus giving way to flaccid paralysis, as in the case of the clinical pictures of these patients during childhood. Then comes a phase in which new axonal buds appear, reincarnating the affected muscle fibers and restoring all or part of their function. Not all axonal buds have complete stability; for this reason, they begin to die after a few years, generating new denervation of muscle fibers and the onset of PPS symptoms. 12
Furthermore, persistent inflammatory changes since the presentation of the initial picture during childhood have been described, considering that there has been an increase in several cytokines, mainly proinflammatory such as INF-γ and TNF, both in the spinal cord and in the CSF of patients with PPS.
Recent proteomics research show increased concentrations of protein fragments and proteins related to inflammation and cell death mechanisms, such as apoptosis, in the CSF of patients with PPS. A possibility is raised of a late aberrant response to the original infection or a persistent immune response due to the persistence of viral particles or an immune response to a neurode-generative process caused by other factors. 3,20 Although there are no major paraclinical contributions, the evolution, symptomatology and chronic pain of the patients tell the story of an event that develops over the years and that should not go unnoticed in patients who have a history of poliomyelitis in their childhood.
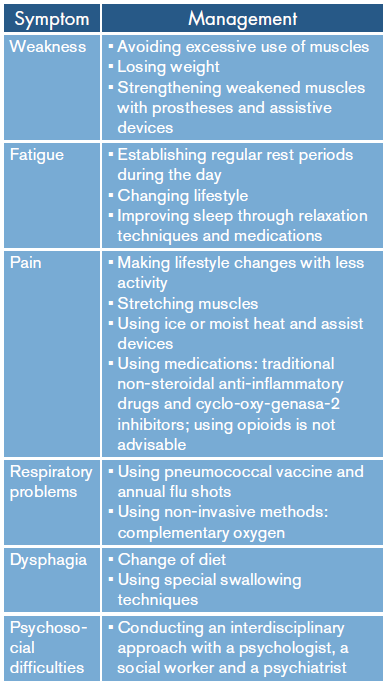
Regarding management, an interdisciplinary approach is suggested, including a neurologist, a rehabilitator, a physiotherapist, an occupational therapist and nursing professionals with experience in rehabilitation of patients with the symptoms related to this syndrome and in psychiatric aspects to allow them to establish management for long-term disability (Table 1). 21-25
Finally, the rehabilitation phase should be considered, especially if it is carried out during the acute phase of the disease. When rehabilitation is carried out late, an evaluation should be made that includes muscle, orthopedic, trophic, respiratory and functional examination. The techniques are chosen according to the findings. The duration of the rehabilitation should be adapted to a precise objective and it is only extended in particular cases. 26
CONCLUSIONS
PPS is a clinical entity difficult to diagnose and treat that requires an interdisciplinary plan to achieve improvement of the initial symptomatology. Based on the fact that management is symptomatic, an improvement in the patient's lifestyle should be achieved to secure a good quality of life. In order to diagnose this entity, the person's childhood history must be kept in mind and the medical history of the past year should be analyzed to determine if the symptomatology has prevailed over time. Although it is incurable, prognosis is positive as long as good therapeutic and symptomatic management is carried out.
Current studies seek to determine if there is an immunological component in PPS, since evidence has shown a correlation with the immune response to inflammation around motor neurons or muscle fibers. This opens a window for young researchers to propose projects that allow searching for alternatives for this type of clinical entities.

