











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La acidosis tubular renal se refiere a un grupo heterogéneo de enfermedades en las que, a pesar de haber una tasa de filtración glomerular (TFG) relativamente normal, se produce acidosis metabólica por un defecto en los túbulos renales, lo que altera su capacidad de regulación del estado ácido-base normal1. En este grupo de enfermedades se presenta acidosis metabólica con anión gap (AG) normal (hiperclorémica), la cual es secundaria a pérdidas de bicarbonato, principalmente en los túbulos proximales, o por defectos en la excreción del cloruro de hidrógeno o de algunas sales que se metabolizan a cloruro de hidrógeno (como el cloruro de amonio) a nivel distal de la nefrona1,2. Este desorden puede ser primario, secundario, adquirido o hereditario, y en ocasiones se asocia a otras enfermedades sistémicas3-8.
Se han definido tres formas principales de acidosis tubular renal: Distal (tipo 1), proximal (tipo 2) e hipercalémica (tipo 4); el tipo 3 corresponde a una forma mixta distal y proximal9-11.
En la acidosis tubular renal tipo 1 (ATR1) se altera la excreción de ácido en el túbulo distal12 y en ausencia de terapia alcalinizante se genera una acumulación progresiva de hidrógenos en plasma que lleva a la disminución del pH plasmático y está acompañada de pH urinario >5.55,6,12. Asimismo, la ATR1 puede acompañarse de hipocalemia secundaria a pérdida de potasio por la acidemia13. En Colombia se desconoce su incidencia debido a la falta de registros, pero en España, con una población de cerca de 45 millones de habitantes, se han registrado 50 casos en Renaltube de ATR hereditaria; de estos, solo 20 son españoles5,6,9; de igual forma, en la mayoría de países europeos la prevalencia es baja5,6,11; en Inglaterra y Francia los estudios genéticos estiman una relación de 1 caso por millón de habitantes1,3,8. Según varias investigaciones, la población con ATR1 se concentra en los inmigrantes de origen árabe8,9,13.
El presente artículo presenta uno de los primeros reportes de compromiso familiar por ATR1 en Colombia; aquí se identifican tres hermanos con esta patología y con una presentación típica autosómica recesiva. Dada la importancia del tema, también se hace revisión de la literatura sobre esta enfermedad.
Presentación del caso 1
Paciente masculino que ingresó por primera vez a un hospital de III nivel de complejidad a los 12 años de edad. Su madre llevaba una historia clínica extrainstitucional que evidenciaba diagnóstico de ATR1 y reportó que tenía otros dos hijos, ambos menores, que presentaban la enfermedad.
El niño presentaba antecedente de hiperplasia pilórica durante sus primeros días de vida, por lo que requirió manejo quirúrgico antes de cumplir su primer mes de edad. Posteriormente desarrolló enfermedad por reflujo gastroesofágico (ERGE), por lo que recibió manejo medico durante los siguientes años. Al momento de llegar a la institución consumía antieméticos y antisecretores.
A los 10 meses de vida, el servicio de gastroenterología lo remitió a nefrología por sospecha de patología renal debido a persistencia de emésis y evidencia de alteración en la función renal. Luego de algunos estudios, nefrología diagnosticó ATR1 y estableció un plan de manejo y seguimiento. A los 16 meses se le detectó nefrocalcinosis y por medio de audiometrías realizadas a los 4 y 12 años de edad se descartaron alteraciones auditivas.
Asimismo, en sus primeras valoraciones extra-institucionales el menor presentaba un déficit en el crecimiento que se corrigió con terapia alcalinizante. Al hospital de III nivel de complejidad llegó con manejo con polietilenglicol (PEG) 7 g/día por estreñimiento crónico, por lo demás tenía control sintomático adecuado.
Al momento de su primera valoración en el servicio de nefrología pediátrica intrainstitucional, el paciente se encontraba en manejo con solución de Shohl (1 litro = ácido cítrico 140 g + citrato de sodio 70 g + citrato de potasio 70 g) 5 cm3 vía oral cada 8 horas y asintomático. Al examen físico se registró peso de 40 kg (percentil 41), talla de 144 cm (percentil 14) y signos vitales dentro de límites normales para la edad. La madre entregó paraclínicos tomados 1 mes antes a la altura de Bogotá, Colombia, que mostraban gases venosos (GV) con pH: 7,383; PCO2: 38,4; HCO 3 22,4; BE: -2,3; BUN: 8,7; creatinina: 0,45 (TFG: 176 mL/min/1 ,73 m 2), calciuria: 70,9 mg/día (1,7 mg/kg/día) y uroanálisis con pH=6, con resto de parámetros normales. Asimismo traía una ecografía de la misma fecha que mostraba riñón derecho de 100x37 mm y riñón izquierdo de 87x46 mm (índice de Hodson 10,8), sin otras alteraciones.
Posteriormente, el paciente fue valorado por el servicio de genética, que solicitó consulta internacional para mapeo genético. Este estudio fue realizado por Renaltube (España) y sus resultados descartaron la presencia de las mutaciones de ATP6V0A4, la más frecuentemente asociada a ATR1, sin que se pudieran descartar otras no descritas en la literatura.
En uno de los controles institucionales por nefrología pediátrica se evidenció que la madre había suspendido el tratamiento con solución de citratos un mes antes; sin embargo, el paciente tenía gases arteriales a la altura de Bogotá, electrolitos séricos y uroanálisis totalmente normales. Por lo tanto, se consideró posible remisión, no se continuó manejo y se solicitaron exámenes de control. En el último control, 8 meses después, el niño se encontraba asinto-mático con signos vitales y examen físico normales y los paraclínicos solicitados mostraron GV con pH: 7,381; PCO2: 47; HCO3: 21; BE: 3, uroanálisis con pH=6 y electrolitos en rangos normales. Con esto se corroboró ATR1 superada y se decidió continuar sin terapia alcalina.
Presentación del caso 2
Paciente masculino con historia clínica extra-institucional que evidenció que, además de ATR1, presentaba diagnóstico de deficiencia de mevalonato quinasa asociada a hiperinmunoglobulinemia D.
Durante sus primeros dos años de vida presentó cuadros bronco-obstructivos a repetición por los que requirió manejo intrahospitalario en múltiples ocasiones, recibiendo eventualmente diagnóstico de asma.
En uno de sus episodios bronco-obstructivos a los 10 meses de edad, el paciente requirió hospitalización y manejo con corticoides; en esa ocasión, a través de resultados paraclínicos, se evidenció un déficit hidroeletrolítico persistente. Teniendo en cuenta estas alteraciones electrolíticas y el diagnóstico reciente, en aquel entonces, de ATR1 en su hermano mayor, se sospechó diagnóstico de acidosis tubular renal. Por lo tanto, al egreso el menor fue remitido al servicio de nefrología pediátrica extrainstitucional con solicitud de estudios de extensión. Al año de edad recibió tal valoración, confirmándose con estudios pertinentes el diagnóstico de ATR1. Desde entonces, ese servicio estableció manejo con solución de citratos y seguimiento, el cual se ajustó progresivamente para lograr metas terapéuticas. Sin embargo, se evidenciaron cuadros eméticos a repetición.
El niño fue valorado por el servicio de gastroenterología pediátrica extrainstitucional donde se estableció diagnóstico de ERGE y se dio manejo antisecretor ocasional con mejoría sintomática. Asimismo, a los 7 años se le detectó nefrocalcinosis y se le descartaron alteraciones auditivas con audiometría. Al igual que su hermano mayor, este paciente también tuvo talla baja durante sus primeras valoraciones extra-institucionales, lo cual se corrigió, y no presentó repercusiones adicionales durante seguimientos extrainstitucionales por alrededor de un año y medio más.
Desde los 6 años de edad comenzó a presentar múltiples episodios de fiebre, artralgias, cefalea esporádica, aparición de aftas y astenia, para lo cual recibió diferentes diagnósticos y tratamientos por 6 meses, hasta que finalmente se sospechó inmunodeficiencia sobreagregada, por lo cual fue remitido a reumatología e infectología pediátrica extrainstitucionales, quienes hicieron estudios de extensión y corroboraron el diagnóstico de deficiencia de mevalonato quinasa asociada a hiperinmunoglobulinemia D a sus 8 años de edad. Desde entonces se inició manejo con lovastatina y colchicina para modular el proceso inflamatorio y prevenir el desarrollo de amiloidosis renal.
A los 9 años de edad fue llevado por primera vez a la institución para manejo multidisciplinario, siendo valorado inicialmente por nefrología pediátrica. El paciente se encontraba en manejo con solución de Shohl 5 cm3 vía oral cada 12 horas, lovastatina y colchicina. Al examen físico se le registró peso de 28 kg (percentil 37), talla de 128 cm (percentil 13) y signos vitales dentro de límites normales para la edad. Los últimos paraclínicos que traía la madre mostraban GV (a la altura de Bogotá) con pH: 7,363; PCO2: 41,9; HCO : 23,3; BE: -2,1, BUN: 6,7, creatinina: 0,4 (TFG: 132 mL/min/1,73 m2), calciuria: 55,6 mg/día (1,98 mg/kg/día) y calcio sérico en 9,35. Traía también una ecografía realizada una semana antes al control en la que se evidenciaba riñón derecho de 98x40 mm y riñón izquierdo de 98x4 5mm (índice de Hodson 9,9), sin hallazgos positivos adicionales.
Posteriormente, el menor fue valorado por el servicio de reumatología pediátrica institucional, que consideró que el paciente se podría beneficiar de la terapia biológica con etanercept para cuadro de deficiencia de mevalonato quinasa asociada a hiperinmunoglobulinemia D. Más adelante, el servicio de genética solicitó secuenciación del gen MVK por riesgo de retinitis pigmentosa dado cuadro autoinflamatorio descrito y envió muestras para mapeo genético en busca de mutaciones de ATP6V0A4 compatibles con de ATR1 cuyo resultado no se conocía al momento de la redacción de este artículo.
La madre suspendió la solución de citratos un mes antes del control por nefrología. Dentro de los paraclínicos de control se registró GV (a la altura de Bogotá) con pH: 7,356; PCO2 44: HCO 3 23,2; y electrolitos séricos y uroanálisis dentro de límites normales. También traía una nueva ecografía y audiometría realizadas tres meses antes de dicho control que no mostraban alteraciones. Se consideró posible remisión de la enfermedad, por lo que no se continuó manejo y se solicitaron exámenes de control. En su último control, 8 meses después, los paraclínicos mostraron gases arteriales (a la altura de Bogotá) con pH: 7,317; PCO 2: 50,9; HCO 3 26,1 ; BE: -0,1; BUN: 19,2, creatinina: 0,62 (TFG: 93,3 mL/ min/1,73 m2), calciuria120 mg/día (3,2 mg/kg/día) y uroanálisis con pH=6; también se le realizó una ecografía renal en la que no se encontró litiasis y en la que los electrolitos fueron normales. El servicio de nefrología consideró estabilidad para ATR1 con base en los paraclínicos previos, la ausencia de síntomas y signos patológicos y la remisión del hermano, por lo que continuó el manejo médico instaurado por reumatología sin adicionar terapia alcalinizante.
Presentación del caso 3
Paciente femenina en cuya historia clínica extra-institucional se evidenció, al igual que sus hermanos, diagnóstico de ATR1.
Esta paciente no presentó complicaciones inmediatas durante sus primeros días de vida. Sin embargo, la madre, teniendo en cuenta el diagnóstico de ATR1 en sus dos primeros hijos, decidió llevarla al mes de vida a valoración por nefrología pediátrica extra-institucional para descartar la presencia de esta enfermedad. Los paraclínicos realizados revelaron alteraciones compatibles con tubulopatía similares a las de sus hermanos. Posteriormente se le confirmó diagnóstico de ATR1 y se estableció seguimiento sin instaurar terapia de suplementación alcalina inicialmente. Sin embargo, durante los siguientes 6 meses presentó cuadros eméticos a repetición y paraclínicos control con alteraciones respecto a iniciales, por lo que se inició manejo con solución de citratos.
Durante los seguimientos se detectaron nefrocalcinosis mediante ecografías renales realizadas en su período de lactante y a los 5 y 6 años de edad. Asimismo, se descartaron alteraciones auditivas con audiometrías normales a los 3 y 8 años, Debido a detección de nefrocalcinosis, su manejo se fue ajustando progresivamente para cumplir objetivos terapéuticos. La talla estuvo constantemente por debajo de 1 desviación estándar para la edad, sin llegar a presentar talla baja como tal.
Esta paciente recibió valoración extra-institucional por el servicio de genética, quienes, teniendo en cuenta contexto clínico e historia familiar, solicitaron estudio genético específico, que al igual que en el hermano mayor fue realizado en Renaltube y tampoco evidenció mutaciones usuales del gen ATP6V0A4 compatibles con ATR1, de nuevo sin poderse excluir otras mutaciones. Los síntomas gastrointestinales persistieron y posteriormente tuvo diagnóstico de ERGE y estreñimiento crónico.
La paciente fue valorada por primera vez en la institución a sus 7 años de edad, a donde llegó con manejo antiemético y antisecretor para ERGE, y con PEG 7 g/día para el estreñimiento, con adecuado control sintomático para el último.
En su primera valoración por el servicio de nefrología pediátrica institucional, la niña se encontraba en manejo con solución de Shohl 13 cm3 vía oral cada 8 horas y presentaba síntomas gastrointestinales dados por dolor abdominal epigástrico tipo ardor, pirosis, disfagia y rumiación. Al examen físico se le registró peso de 25 kg (percentil 49), talla de 123 cm (percentil 26) y signos vitales dentro de límites normales para la edad. La madre traía paraclínicos que habían sido tomados 1 semana antes y que evidenciaban GV (a la altura de Bogotá) con pH: 7,432; PCO 2: 31,1; HCO3: 20,3; BE: -2,8; BUN: 6,4; creatinina: 0,37 (TFG: 137 mL/min/1,73 m2), calciuria: 248,5 mg/día (9,7 mg/kg/día), así como una ecografía de la misma fecha que mostraba riñón derecho de 93x33 mm y riñón izquierdo de 91x43 mm (índice de Hodson 9,6), sin otras alteraciones. Por lo anterior se ajustó la terapia aumentando solución de citratos 15 cm3 cada 8 horas e intentando manejo con dieta hiposódica y restricción en aporte de calcio.
Posteriormente, la paciente fue valorada por el servicio de gastroenterología pediátrica, quienes consideraron exacerbación de los síntomas de ERGE y estreñimiento no controlado, por lo que solicitaron estudios de extensión y ajustaron manejo con PEG 15 g al día e indicaron aumento en aporte hídrico oral.
Durante un año y medio de seguimiento, nefrología pediátrica ajustó el manejo con terapia alcalina y medidas dietéticas y un mes antes de la cita de control la madre de la paciente suspendió el tratamiento. Los resultados de los paraclínicos sin medicación fueron GV (a la altura de Bogotá) con pH: 7,344; PCO2: 43; HCO3: 23; BE: -2,5 y electrolitos séricos y uroanálisis normales. Al igual que con sus hermanos, se consideró posible ATR1 superada, por lo que no se continuó manejo y se solicitaron exámenes de control.
Ocho meses después, en el último control, la niña se encontraba asintomática, con desarrollo adecuado y con GV (a la altura de Bogotá) con pH: 7,24; PCO2: 63; HCO3: 27; BE: -1,6, electrolitos sin alteraciones, y parcial de orina (PO) con pH 5. Sin embargo, al igual que en el hermano, se consideró cuadro de ATR1 estable, orina con adecuada acidificación y concentración, por lo que decidió continuar sin manejo hasta el próximo control para evaluar evolución.
Es importante mencionar que ninguno de los tres hermanos tuvo antecedentes adicionales de importancia, que ni los embarazos y ni los partos tuvieron complicaciones y que todos nacieron a término. La familia es de raza mestiza, natural y procedente de Duitama (Boyacá) y ha residido siempre en adecuadas condiciones económicas en un hogar con todos los servicios básicos. Los padres, a quienes no se les realizaron estudios genéticos de extensión, se encontraban aparentemente sanos, negaban consanguinidad, no presentaban bajo peso o estatura de modo constitucional, no tenían antecedentes de importancia ni habían presentado cuadros patológicos que sugirieran alteraciones renales como las observadas en sus hijos.
Acidosis tubular renal tipo 1 (distal)
Fisiopatología
En el túbulo distal la excreción de iones de hidrógeno a la luz tubular la realizan las células intercaladas (tipo A), presentes en la región final del túbulo contorneado distal y en el túbulo colector (Figura 1)13. El H2 O intracelular se disocia en iones H+ y OH-; los primeros son secretados a la luz tubular por medio de las bombas H+-ATPasa y H+-K+-ATPasa y los segundos se combinan con CO para formar HCO3 - en una reacción catalizada por la anhidrasa carbónica II (ACII). El HCO3 - pasa al espacio peritubular a través del intercambiador de aniones (AE1), que permite la entrada de Cl- por un mecanismo de contratransporte con HCO3-.

Fuente: Elaboración con base en Batlle & Haque6.
Figura 1 Rol de la célula? intercalada en el mantenimiento del equilibrio ácido-básico.
De este modo, en la ATR1 distal la disminución en la secreción de H+ se puede producir por disminución en la actividad neta de la bomba de protones o por aumento en la permeabilidad de la membrana luminal al H+13-16.
La disminución en la actividad neta de la bomba de protones es el principal mecanismo productor de ATR1 y puede deberse a varias alteraciones que la afectan directa o indirectamente16-18. Por ejemplo, se han encontrado varias mutaciones en genes que codifican subunidades de la bomba H+-ATPasa y que conllevan a una pérdida en su función de secreción de protones8,19,20. Por otro lado, en el síndrome de Sjögren (SS) con ATR1 se ha evidenciado ausencia completa de la bomba H+-ATPasa, la cual se da por mecanismos inmunológicos poco entendidos21,22. Asimismo, en este síndrome se han identificado títulos altos de auto-anticuerpos contra la ACII que disminuyen su actividad, la generación de iones de H+ en la célula intercalada y la secreción por la bomba de protones23. Finalmente, se han identificado mutaciones en AE1 en varias familias con formas hereditarias de ATR1, que al disminuir la actividad de este transportador llevarían a acumulación de HCO3- en la célula A con consecuente disminución en la generación de H+ intracelular24-28.
Un mecanismo de producción de ATR1 menos frecuente se relaciona con la permeabilidad de la membrana luminal al H+. Dado que la concentración de H+ muchas veces es mayor en la orina que en el espacio extracelular, esta membrana debe ser relativamente impermeable para impedir que estos iones regresen a las células tubulares y posteriormente a la circulación sistémica. Cuando la permeabilidad de esta membrana disminuye, como ocurre en la nefrotoxicidad asociada al uso de anfotericina B, estos iones tienden a regresar al espacio extracelular, lo que lleva a acidosis metabólica hiperclorémica29,30.
Etiología
En niños, la ATR1 casi siempre tiene origen primario, identificándose varias mutaciones genéticas que se transmiten de forma autosómica dominante o autosómica recesiva. No obstante, en cerca del 20 % de los casos no se logra identificar alguna mutación conocida1,5,19,31. En la Tabla 1 se distinguen las tres formas genéticas principales de ATR1 primaria.
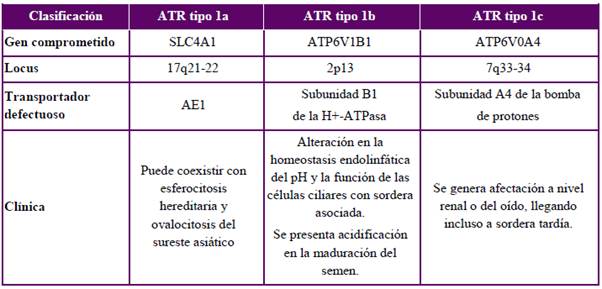
Además de los desórdenes genéticos descritos, el síndrome de Ehlers-Danlos y la anemia de células falciformes también pueden ser una etiología genética de ATR1, aunque de tipo secundaria43,44.
Algunas causas secundarias adquiridas y menos frecuentes en niños son el consumo de algunos medicamentos y los desórdenes autoinmunes. Dentro de los primeros destaca la anfotericina B, que puede producir ATR1 irreversible29,30; el litio, que puede generar una forma incompleta de ATR1 en la que hay disminución en acidificación de la orina a pesar de que el pH plasmático se encuentre normal45, y la isofosfamida46. Como desórdenes autoinmunes asociados se han reportado el SS y el lupus eritematoso sistémico, que pueden producir este desorden por mecanismos inmunológicos aún desconocidos21-23,47,48.
Manifestaciones clínicas
Las manifestaciones varían considerablemente según la etiología. En el caso de las formas primarias de ATR1 se presentan cuadros más estereotipados en edades más características.
La ATR1 recesiva es la más frecuente y de mayor severidad; esta cursa con acidosis metabólica hiperclorémica severa e hipocalemia moderada a severa19,49-51. Por lo tanto, sus manifestaciones suelen presentarse de forma temprana y el diagnóstico suele realizarse más precozmente en relación a la forma dominante de la enfermedad19,49,50,52.
La Figura 2 resume las manifestaciones clínicas de la ATR1 de acuerdo a sistemas de órganos comprometidos, siendo presentadas principalmente en la forma recesiva de la enfermedad.

Fuente: Elaborado con base en53-66.
Figura 2 Manifestaciones clínicas de la acidosis tubular renal distal de acuerdo a sistemas de órganos comprometidos.
Los pacientes con la forma recesiva de ATR1 presentan dolores abdominales o lumbares secundarios a nefrocalcinosis y/o litiasis renal7,50 que se explican por la orina alcalina que favorece la precipitación de los cristales de fosfato de calcio54. Por otra parte, existe disminución en la concentración de citrato urinario (potente inhibidor en la formación de cálculos de calcio), lo que favorece la precipitación de este último mineral55,56. Además, la acidemia promueve la hipercalciuria al aumentar la liberación de fosfato de calcio del hueso por el sistema de búfer óseo y al disminuir directamente la reabsorción tubular de estos minerales57,58.
Las anteriores alteraciones son frecuentes en los pacientes con ATR1 de tal forma que un diagnóstico tardío puede comprometer el tamaño de los riñones o llevar incluso a insuficiencia renal terminal, por lo que se recomienda la realización de ultrasonografía renal anualmente durante el seguimiento6,59.
De otra parte, las alteraciones auditivas neurosensoriales se presentan exclusivamente en la forma recesiva de ATR137,42,49. Sin embargo, las formas recesivas de ATR1 no necesariamente se acompañan de sordera, además las alteraciones auditivas asociadas presentan una considerable heterogeneidad fenotípica20,41,42. En situaciones donde haya disponibilidad de estudios genéticos, el estado auditivo del paciente no debe influir a priori en una decisión sobre qué genes estudiar, y el hallazgo de mutaciones asociadas a alteraciones auditivas neurosensoriales obliga a la realización de exámenes auditivos periódicos20,37,49,60.
En el caso de la ATR1 de herencia dominante, las manifestaciones suelen ser leves, siendo la nefrocalcinosis y la litiasis renal las más frecuentes7,50, y pueden cursar con acidosis metabólica leve o no (ATR1 incompleta), así como normocalemia o hipocalemia leve a moderada25,50. Rara vez se presentan enfermedades óseas o retraso del crecimiento25,50 y tampoco son habituales otras alteraciones descritas para la ATR1 recesiva. Por lo tanto, la forma dominante de ATR1 suele ser descubierta tardíamente, incluso en la adultez50,67.
Diagnóstico
La Figura 3 ilustra el abordaje inicial ante la sospecha clínica de acidosis tubular renal.

Para diagnosticar un posible caso de ATR1 debe valorarse la excreción urinaria de NH4+, que por disminución en la secreción de H+ a la luz tubular en ATR1 siempre se encuentra disminuido (<2040 mEq/día) 17,69,74. Esta medida es de particular importancia en el diagnóstico diferencial de la acidosis metabólica secundaria a inhalación de tolueno, que si bien presenta AG normal e hipocalemia como en la ATR1, cursa con excreción de amonio normal o elevada (>40 mEq/día) 75. Dado que muy pocos laboratorios pueden medir directamente el amonio, el AG urinario (AGu) ha surgido como una medida indirecta del NH4+ en orina74. La Figura 4 ilustra conceptos para entender el uso del AGu.

Según esto, el AGu se haría más negativo a medida que la concentración de NH4+ aumentara, lo cual incrementaría simultáneamente el Cl. En algunos estudios se evaluó la relación entre AGu y concentración urinaria de NH4+, encontrándose relación lineal inversa y desarrollándose la fórmula [NH4+ = -0.8AGu + 82] para la estimación del NH4+74,76.
Asimismo, en estos estudios se promedió el AGu de personas con función tubular normal entre -20 y -50 mEq/L y se evidenciaron valores mayores (menos negativos) en pacientes con excreción de NH4+baja74,76. Por lo tanto, en la ATR1 se encontraría un AGu elevado, generalmente positivo, y un NH4+ disminuido, bien sea estimado o medido directamente17,69.
Tratamiento
La corrección de la acidemia ha demostrado gran eficacia en la ATR1, pues disminuye la pérdida renal de potasio, restaura el crecimiento normal, retrasa la nefrocalcinosis y la litiasis urinaria, previene la progresión hacia insuficiencia renal crónica e incluso disminuye las alteraciones óseas asociadas a esta enfermedad77-80. La terapia alcalinizante es por tanto el manejo indicado para ATR1, ya que busca alcanzar niveles plasmáticos relativamente normales de bicarbonato.
El bicarbonato de sodio y el citrato de sodio son alternativas viables; sin embargo, el primero genera ocasionalmente una intolerancia a nivel digestivo que constituye uno de los principales obstáculos81. Las dosis requeridas en niños pueden llegar hasta 4-8 mEq/kg/día, mientras en adultos 1-2 mEq/kg/día suelen ser suficientes1,82. El citrato de potasio es también una excelente alternativa, principalmente por su utilidad para la reposición de K+ y tolerabilidad83; además, el citrato aportado al filtrarse en el riñón puede aumentar directamente la excreción urinaria de citrato, si bien el principal mecanismo para este aumento es la disminución de la acidemia, causante de la reabsorción tubular aumentada de citrato en la ATR155,56,78,79.
La ATR1 es casi siempre una enfermedad permanente, por lo que la terapia con álcalis debe ser continuada de por vida84. Cuando se diagnostica de manera precoz, el paciente puede tener menos complicaciones y su pronóstico mejorará1,85. Así mismo, una dieta hiposódica podría tener efectos benéficos, por depleción leve de volumen, aumentando la reabsorción de Na+ a nivel tubular proximal y, de forma secundaria, la de HCO3-86,87.
Discusión
Algunas características del cuadro presentado en los tres pacientes llaman la atención y merecen ser analizadas a la luz del conocimiento actual sobre ATR1:
Los tres hermanos presentaron distintas comorbilidades desde los primeros meses de vida; sin embargo, la manifestación inicial de la ATR1 en los tres hermanos consistió en síndrome emético refractario a corta edad, lo cual se reporta frecuentemente en la literatura sobre ATR17,88,89. Los pacientes también tuvieron desarrollo temprano de nefrocalcinosis y talla baja, aunque la hermana menor solo presentó mediciones en rango de riesgo de talla baja, siendo todos hallazgos clínicos comunes6,7,11,50. Estos casos también coincidieron con lo reportado en la literatura respecto a las alteraciones clínicas que se manifiesten a edades muy tempranas en las presentaciones hereditarias recesivas de ATR119,49,50,52.
Como se mencionó antes, la terapia temprana suele llevar a un excelente pronóstico1, lo que se reflejó en adecuada evolución y desarrollo de estos pacientes. Así, tal como se ha reportado previamente77-80, tanto la nefrocalcinosis como la talla baja mejoraron con adecuado manejo.
Aunque no se realizó control ecográfico renal anualmente, tal como es recomendado6,59, se hicieron por lo menos cuatro ecografías renales durante el seguimiento, las cuales evidenciaron la remisión de la nefrocalcinosis. Por otra parte, con el tratamiento alcalinizante se mejoraron la calciuria, los gases arteriales y el potasio sérico durante los seguimientos, lo que coincide con lo descrito en la literatura77-80.
Se resalta que en ninguno de los pacientes hay compromiso auditivo, lo que es compatible con ATR1 tipo 1c8. No obstante, los estudios realizados por Renaltube no lograron identificar las principales mutaciones del gen ATP6V0A4 compatibles con esta presentación. Lo anterior no descarta el diagnóstico, pues probablemente se trata de una mutación no descrita hasta el momento en la literatura internacional. Esto no es inusual si se tiene en cuenta que, si bien en población pediátrica la principal causa de ATR1 es genética, hasta en un 20% de los casos de ATR1 primaria en niños no logra identificarse la mutación subyacente1,5,19,31. La descripción de estas mutaciones no es el objetivo principal de esta publicación, sin embargo, su reporte puede ser la base para estudios futuros19,20,37,41,42.
Es curioso que en uno de los hermanos haya presencia concomitante de deficiencia de mevalonato quinasa asociada a hiperinmunoglobulinemia D, una enfermedad metabólica que definitivamente altera el pronóstico y la evolución del paciente. La asociación de ATR1 congénita y enfermedades metabólicas no ha sido claramente descrita en la literatura, por lo que vale la pena informar este evento para que en futuros estudios y reportes de casos se determine algún tipo de relación.
Algo que también llamó la atención fue la persistencia de la mejoría clínica en los tres pacientes luego de la suspensión de terapia alcalinizante y que dentro de las alteraciones bioquímicas derivadas de esta enfermedad solo presentaron disminución en el pH hasta claros rangos de acidemia, pero con uroanálisis, electrolitos séricos y calcio urinario normales. La principal meta en el manejo se mantuvo conseguida en los tres pacientes: la corrección de síntomas asociados a ATR1, la remisión de la nefrocalcinosis y evitar el daño renal permanente1. Debe hacerse la salvedad que aún continúan los seguimientos y que futuros estudios y valoraciones definirán si efectivamente no volverán los requerimientos de terapia alcalina. Otra posibilidad es que las necesidades de terapia alcalina hayan disminuido sin desaparecer del todo, como ha sido claramente descrito a medida que los pacientes con ATR1 van creciendo82, lo que también se confirmará con el seguimiento.
Conclusiones
A pesar de que las manifestaciones clínicas de los casos presentados están en concordancia con lo reportado en la literatura sobre ATR1, esta publicación resalta algunos hechos no comunes que ameritan tenerse en cuenta en investigaciones futuras. Por un lado, la asociación de uno de los casos con deficiencia de mevalonato quinasa con hiperinmunoglobulinemia D y, por el otro, la desaparición de la necesidad de la terapia alcalinizante, particularmente en el primer caso, que tampoco es usual de acuerdo al conocimiento actual de ATR1.
El estudio de familiares de pacientes afectados por esta patología es importante, ya que existen diferentes mutaciones genéticas con diversas formas de transmisión y manifestaciones clínicas asociadas. Se debe realizar entonces una búsqueda activa en familiares, evaluando los factores de riesgo descritos previamente. Si bien en el momento no hay protocolos validados con una adecuada metodología científica de análisis de costo-efectividad para este tipo de estudio, su posibilidad es algo que debe contemplarse e investigarse en estudios futuros.
Llama la atención no haber evidenciado en estos pacientes mutaciones previamente asociadas a ATR1. Sin embargo, existe la posibilidad de que hayan presentado mutaciones no descritas que deberán estudiarse en el futuro.
Este tipo de patología en pediatría debe ser estudiada y manejada de forma multidisciplinaria con nefrología pediátrica, pediatría general, genética, endocrinología pediátrica e incluso nutrición, y se debe establecer un plan de seguimiento periódico que incluya consejería genética cuando los resultados de los estudios así lo indiquen.

