











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
La hipoacusia tiene una incidencia de uno a dos por mil nacidos vivos. Se estima que del 60% al 80% de las hipoacusias congénitas o prelinguales son de causa genética, y que existe gran heterogeneidad de locus y alélica, así como expresividad variable. El 70% de estas corresponde a sordera no sindrómica, en el que la hipoacusia es el único hallazgo clínico, y que en su gran mayoría son ocasionadas por variantes patogénicas en un solo gen y cuyos patrones de herencia pueden ser autosómico recesivo (70 a 80%), autosómico dominante (10 a 20%) y ligada al cromosoma X, con una aproximación del 1% al 5%. A la fecha se han descrito 203 tipos o loci de sordera congénita no sindrómica y 133 genes relacionados [1-5]. Los genes más frecuentemente involucrados son el GJB2, en primer lugar, y el SLC26A4, en segundo. De este último se han reportado varias variantes patogénicas asociadas a sordera en Turquía, Irán y México [6]. De las sorderas no sindrómicas ligadas al X, a la fecha se han identificado siete loci o tipos (DFNX1 - 7) [1,2].
Malformación del Oído Interno (MOI)
El oído interno tiene una anatomía compleja y varios genes están involucrados en su desarrollo, especialmente entre las ocho y diez semanas de gestación, por lo que se ha descrito un espectro amplio de malformaciones que ocasionan sordera neurosensorial [7]. Del 20% al 40% de las hipoacusias neurosensoriales congénitas, ya sean sindrómicas o no, presentaron MOI cuando fueron estudiadas con métodos de imagen como la tomografía computarizada (TC) y la resonancia magnética (RM). Estas malformaciones incluyen la combinación de alteraciones en la estructura ósea y membranosa. Existen diversas clasificaciones de las MOI; la más aceptada es la de L Sennaroğlu [5,8-10]
Las hipoplasias cocleares representan del 15% al 23% de las MOI y se clasifican como tipos 1 a 4. La arquitectura coclear interna puede ser normal o anormal según el subtipo. Así, los tipos 1 y 2 son versiones pequeñas de una cóclea dividida de forma incompleta, mientras que los tipos 3 y 4 son cócleas pequeñas, pero con una estructura interna casi completa [7].
Aplasia laberíntica completa, conocida como aplasia de Michel, es el resultado de la detención del desarrollo de la placoda ótica antes de la tercera semana de gestación y solo supone el 1% de las MOI [10].
Partición incompleta de la cóclea (PI)
Las anomalías de partición incompleta representan un grupo de malformaciones cocleares, en las que existe una clara diferenciación entre cóclea y vestíbulo, con dimensiones externas normales y diversos defectos de arquitectura interna. Las particiones incompletas constituyen el 41% de todas las MOI. Existen tres tipos diferentes, de acuerdo a los defectos modiolares y del septo interescalar [7,8].
Partición incompleta tipo I (PI-I)
Se denominó “malformación quística cócleo-vestibular” [9] a la partición incompleta tipo I (PI-I). En esta anomalía existe una clara diferenciación entre cóclea y vestíbulo. La cóclea se localiza en la parte anterolateral del fondo del conducto auditivo interno (CAI), carece de modiolo y septo interescalar, dando la apariencia de una estructura quística vacía. Además, puede acompañarse de dilatación vestibular, con un acueducto vestibular normal. Se presenta en aproximadamente el 20% de todas las MOI [7-9].
Partición incompleta tipo II (PI-II)
Se trata de la anomalía clásica de Mondini, que se caracteriza por la triada: ápice coclear quístico con giros basales normales, vestíbulo agrandado y acueducto vestibular agrandado (EVA). Se presenta en el 19% de todas las hipoacusias neurosensoriales (HNS) congénitas no sindrómicas y se ha asociada al gen SLC26A4, uno de los diez más frecuentes en la sordera no sindrómica. Algunos autores la reportan como la segunda causa de sordera no sindrómica. El fenotipo más frecuente encontrado en las mutaciones SLC26A4 es acueducto vestibular amplio y PI-II [6-8].
Partición incompleta tipo III (PI-III)
Es una MOI poco frecuente, [10-12] y se puede distinguir la cóclea y el vestíbulo. Presenta un septo interescalar con un modiolo ausente, las medidas de la cóclea son normales, tiene un aspecto quístico, sin arquitectura interna, y el vestíbulo se encuentra dilatado. La cóclea se localiza directamente lateral al conducto auditivo interno (CAI), el cual es “bulboso” o dilatado, con una separación incompleta de las espirales de la cóclea, y ambos están ampliamente comunicados por una deficiencia completa de la lámina cribosa. La cápsula ótica es más delgada alrededor del laberinto membranoso, en comparación con una cóclea normal [7,9-12]. La PI-III es una de las MOI menos frecuentes; también, entre los grupos de partición incompleta. Se observa en el 2% de todas las malformaciones cocleares, de herencia ligada al cromosoma X. Típicamente afecta a los hombres y cursa con hipoacusia mixta rápidamente progresiva [10-13]. Se ha asociado frecuentemente con una variante patogénica del gen POU3F4, y en algunos casos variantes del gen COL4A6 [7,8,10-13].
Sordera de causa Ligada al cromosoma X (LX)
Cuando ocurre una sordera ocasionada por una variante patogénica en un gen localizado en el cromosoma X, hablamos de sordera ligada al X (LX). Representa aproximadamente del 1% al 5% de los casos de sordera sindrómica y no sindrómica. Clásicamente se mencionan dos patrones: herencia ligada a X recesiva (LXR) y herencia ligada a X dominante (LXD).
En la sordera LXR solo se observan varones afectados; las mujeres portadoras obligadas no tienen sordera, pero el 50% de sus hijos varones tendrán riesgo de ser sordos y el 50% de sus hijas tendrán riesgo de ser portadoras. Los varones afectados con un gen de sordera LXR (hemicigotos) nunca podrán transmitir el gen a un hijo varón, pero todas sus hijas, usualmente sanas oyentes, serán portadoras.
La sordera LXD afecta más a las mujeres que a los hombres, pero suele ser más severa en varones. La herencia es dominante cuando se expresa en estado de heterocigosis, es decir, en presencia de un solo alelo con la variante patogénica. Las mujeres heterocigotas con sordera LXD trasmiten el gen al 50% de sus hijos independiente del sexo, teniendo la probabilidad del 50% de ser sordos, ya sean hombres o mujeres. Los varones sordos con LXD, hemicigotos, transmitirán el gen a todas sus hijas, por lo que presentarán el padecimiento. Sus hijos varones serán sanos, libres de la variante patogénica del gen. De la sordera LXD solo se ha descrito un tipo, DFNX4 [1,14-16].
A la fecha solo se han identificado siete loci o tipos de sorderas no sindrómica LX (DFNX1 - 7) y seis genes clonados: PRPS1 para DFNX1, POU3F4 para DFNX2, SMPX para DFNX4, AIFM1 para DFNX5, COL4A6 para DFNX6 y GPRASP2 para DFNX7. De estos, cuatro presentan MOI [1,2,17-20]. En la Tabla 1 se resumen las características clínicas: genéticas, audiológicas y MOI de los siete tipos de sordera no sindrómica LX descritos a la fecha. Esta tabla se construyó y modificó con información de las tablas de G Van Camp, AE Shearer, V Corvino y G Xing. [1,2,17-20].
Tabla 1 Resumen Manifestaciones Clínicas de la Sordera no Sindrómica, LX
| Genética | Audiología | MOI | ||
|---|---|---|---|---|
| Locus / OMIM / Herencia | Gen/ Localización | Presentación | Características de hipoacusia | |
| DFNX1/ 304500/ LX | PRPS1/ Xq22.3 | Postlingual, en quinta década, varones un inicio más temprano que las mujeres. Prelingual congénita | HNS de severa a profunda progresiva, postlingual. HNS congénita | No |
| DFNX2/ 304400/ LXR | POU3F4/ Xq21.1 | Prelingual | HNS / HC / HM, Progresiva, la severidad puede ser hasta profunda. Mujeres portadoras pueden tener HNS leve | PI-III, Hipoplasia de cóclea, sin modiolo, CAI dilatado, Fijación del estribo |
| DFNX3/ 300030/ LX | Xp21.2 | Congénito | HNS severa a profunda bilateral. En mujeres adultas portadoras, HNS de Leve a moderada, de aparición tardía | No |
| DFNX4/ 300066/ LXD | SMPX/ Xp22.12 | Postlingual. En varones inicia de tres a siete años; en mujeres de la niñez hasta la cuarta década | HNS, de media a profunda, progresiva, iniciando en frecuencias altas | No |
| DFNX5/ 300614/ LXR | AIFM1/ Xq26.1 | Postlingual, inicio antes o durante la adolescencia | Neuropatía Auditiva, Neuropatía sensorial periférica tardía en edad adulta, lentamente progresiva. Acúfenos perdida de función de las CPE | Hipoplasia del nervio coclear |
| DFNX6/ 300914/ LXR | COL4A6/ Xq26.1 | Prelingüística/postlingüística | HNS severa, bilateral/progresiva, en mujeres portadoras durante la tercera y cuarta décadas de la vida HNS leve a moderada | Cóclea malformada, separación incompleta de cóclea del CAI bilateral |
| ? DFNX7/ 301018/ XLR | GPRASP2 / Xq26 | Congénita (Nota: solo en una familia china) | HM ó HC bilateral, congénita, puede ser progresiva | CAI dilatado, atresia o estenosis de CAE, anomalías de PA |
Abreviaturas: LX: Ligado a X; LXD: Ligado a X Dominante; LXR: Ligado a X Recesivo; MOI: Malformación de Oído Interno; HNS: Hipoacusia Neurosensorial; HC: Hipoacusia Conductiva; HM: Hipoacusia Mixta; PI-III: Partición Coclear Incompleta Tipo III; CAI: Conducto Auditivo Interno; CPE: Células Pilosas Externas; CAE: Conducto Auditivo Externo; PA: Pabellón auricular.
La sordera ligada al cromosoma X-2 (DFNX2, OMIM 304400), también conocida como sordera conductiva con fijación congénita de la base del estribo, se acompaña de MOI y constituye aproximadamente la mitad de la sordera no sindrómica LX, causada por variantes patogénicas en el gen POU3F4. Este es el primer gen identificado como causante de sordera no sindrómica LX y el más frecuente, cerca del 50%, produciendo diversos fenotipos: hipoacusia neurosensorial (HNS), hipoacusia conductiva (HC), hipoacusia mixta (HM), con severidad profunda, anomalías vestibulares y fijación congénita de la base del estribo y la partición incompleta de la cóclea PI-III. Además de asociarse con el gen POU3F4 [5,11,12], se ha descrito una variante en el gen COL4A6. Sin embargo, esta última se encontró en una sola familia húngara con HNS asociada a MOI. Recién se reportó otra variante de COL4A6 asociada a HNS LX sin MOI; su papel aún no es claro [19]. Las variantes en el gen COCH se han sugerido como posible causa de MOI y una mutación del gen FOXF2 es responsable de la PI- I [5].
Las sorderas no sindrómicas LX son poco frecuentes. En México, hasta donde sabemos, no existe algún informe. Además, son heterogéneas audiológicamente, así como en la presencia o ausencia de MOI, como observamos en la Tabla 1. Es importante identificar las causas genéticas de la HNS congénita y MOI LX para orientar la asesoría genética, que incluye terapéutica. Por ejemplo, auxiliares auditivos o implante cocleares, dependiendo del tipo de malformación y pérdida auditiva; decisiones reproductivas, dependiendo del sexo. En ese sentido, se hace necesario delimitar el fenotipo y reportar casos nuevos.
Siendo tan poco frecuentes las sorderas neurosensoriales, con malformación de oído interno, ligadas X, se considera necesario ampliar su conocimiento e implementar estudios moleculares.
Métodos
Se estudiaron tres generaciones de una familia, en la que existen nueve varones sordos, sin desarrollo de lenguaje.
A todos los individuos participantes se les realizó historia clínica completa por médica genetista, evaluación audiológica por médica audióloga y estudio de imagen por médico radiólogo.
Los individuos estudiados se numeraron o designaron de acuerdo a su posición en el árbol genealógico, iniciando con las generaciones en número romano y su posición en la misma con número arábigo [21].
El estudio de esta familia forma parte del “Proyecto de Investigación de hipoacusia familiar”, aceptado por los comités de Investigación y Ética en Investigación del Instituto Nacional de Rehabilitación Luis Guillermo Ibarra Ibarra (INR LGII), con número de registro 12/13. A todos los participantes involucrados se les informó el objetivo del presente estudio, aceptaron participar voluntariamente y firmaron una carta de consentimiento informado. En el caso de los menores de edad, mayores de ocho años, de igual manera, se les explicó la razón del proyecto y se les exhortó para que participaran, por lo que firmaron la carta de asentimiento informado del menor.
Evaluación audiológica y de imagen
Se realizó audiometría tonal por vía aérea y ósea, previa otoscopia y timpanometría, en cuatro pacientes y una madre. Para la timpanometría (226 Hz) se utilizó el impedanciómetro AT235/ Interacoustics en cámara sonoamortiguada, mediante la colocación de una sonda pediátrica en el conducto auditivo externo -primero derecho y después izquierdo-, para evaluar la curva timpanométrica, y posteriormente se evaluaron los reflejos estapediales ipsilaterales. La audiometría es un estudio subjetivo, que requiere de la colaboración del paciente y que permite identificar el umbral de audición. Se utilizó audiómetro de plataforma Eclipse/Interacoustics en cámara sonoamortiguada. Se procedió a reproducir sonidos puros al paciente a través de audífonos TDH 39 -primero en oído derecho y posteriormente en izquierdo- en las frecuencias de 125, 250, 500, 1000, 1500, 2000, 3000, 4000 y 8000 Hz. En cuanto el paciente detecta los sonidos levantará la mano o bajará ficha (audiometría lúdica), corroborando las respuestas en dos a tres ocasiones. En pacientes usuarios de auxiliares auditivos, se evaluó su ganancia a través de campo libre en las frecuencias de 250 a 4000 Hz. Al paciente de un año de edad se le realizaron Potenciales Evocados Auditivos de Tallo Cerebral (PEATC) y Potenciales Evocados Auditivos de Estado Estable (PEAEE), debido a su edad. [22]
El estudio de imagen se realizó mediante TC de oídos y mastoides. Se aplicaron en cuatro pacientes varones y una madre oyente considerada portadora. Los estudios se clasificaron según Sennanoğlu y cols. [8,9,13].
Estudio clínico genético y banco de ADN
Se estudio la genealogía con catorce individuos, en tres generaciones, de los cuales nueve eran sordos, sin desarrollo de lenguaje, y cinco madres consideradas portadoras. Se tomó muestra de sangre periférica a dieciséis miembros de la familia: ocho varones sordos, cuatro madres oyentes portadoras y cuatro hermanas oyentes, posibles portadoras. Se extrajo ADN mediante técnica estándar, para creación de banco de ADN de la familia, para posterior estudio molecular, por el Dr. Mustafa Tekin del John P. Hussmann Institute for Human Genomics, Department of Human Genetics, Miami, USA.
Resultados
Resultados evaluación audiológica
Todas las otoscopías de los pacientes mostraron hallazgos normales, además de curvas A de Jerger y reflejos estapediales ipsilateralmente ausentes de forma bilateral.
En el paciente IV-3 se encontró por PEATC, HNS media bilateral para tonos altos (Los tonos altos son los que tienen una frecuencia de 2000 a 5000 Hz) [22] (Imagen 1); y con los PEAEE, HNS media a severa derecha y superficial a severa izquierda (Imagen 2).
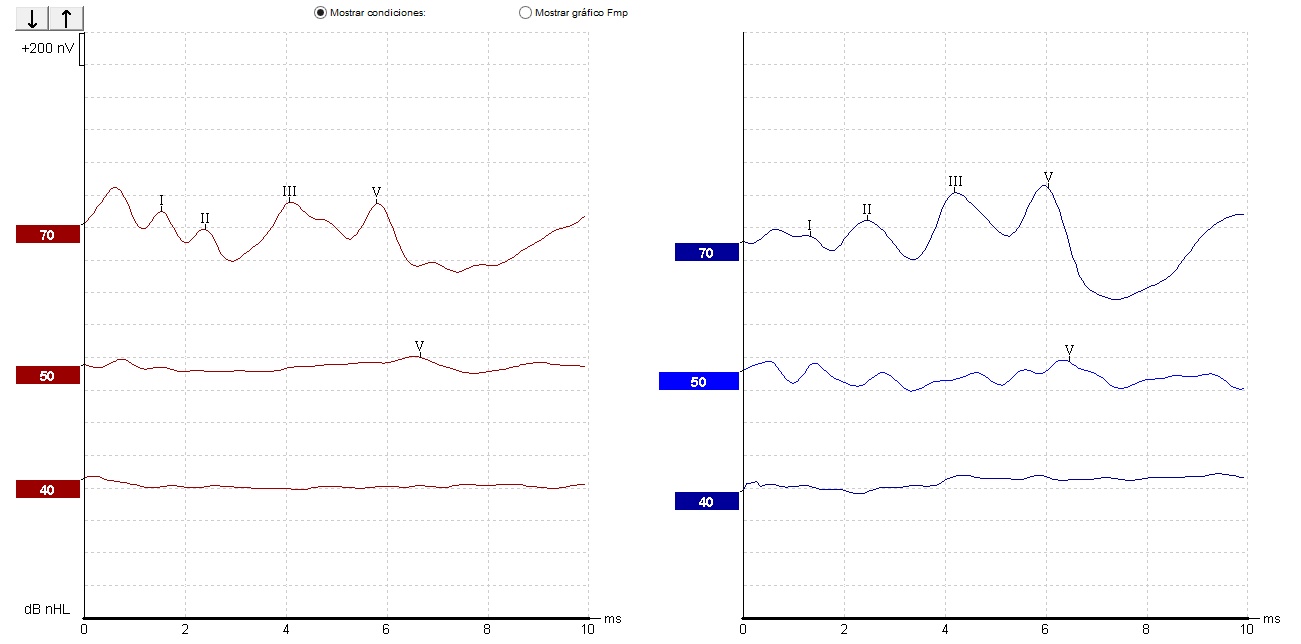
Nota. Se observa umbral de onda V a 50 dB de forma bilateral; datos compatibles con hipoacusia media bilateral para tonos altos
Imagen 1 Potenciales Evocados Auditivos de Tallo Cerebral (PEATC) del paciente IV-3

Nota. Se observan umbrales estimados con curvas de perfil ascendente, compatibles con hipoacusia media a severa derecha y superficial a severa izquierda
Imagen 2 Potenciales Evocados Auditivos de Estado Estable (PEAEE) del paciente IV-3
Los resultados audiológicos de cinco pacientes varones y una madre oyente portadora se muestran en la Tabla 2. Se describieron siguiendo las recomendaciones internacionales para hallazgos audiológicos en sorderas hereditarias [23], encontrando lo siguiente:
Tabla 2 Diagnóstico audiológico
| Individuo | Dx. Audiológico | Dx. MOI | Ver |
|---|---|---|---|
| IV-1 | HNS severa derecha con curva de perfil irregular. HNS profunda izquierda con restos auditivos, congénita | PI-III | Imagen 3 |
| IV-2 | HNS profunda bilateral con curvas de perfil plano, congénita | PI-III | Imagen 4 |
| IV-3 | HNS media a severa bilateral congénita | PI-I | Imagen 1 y 2 |
| IV-4 | HNS profunda bilateral con curva de perfil irregular derecha y curva plana izquierda, congénita | PI-III | Imagen 5 |
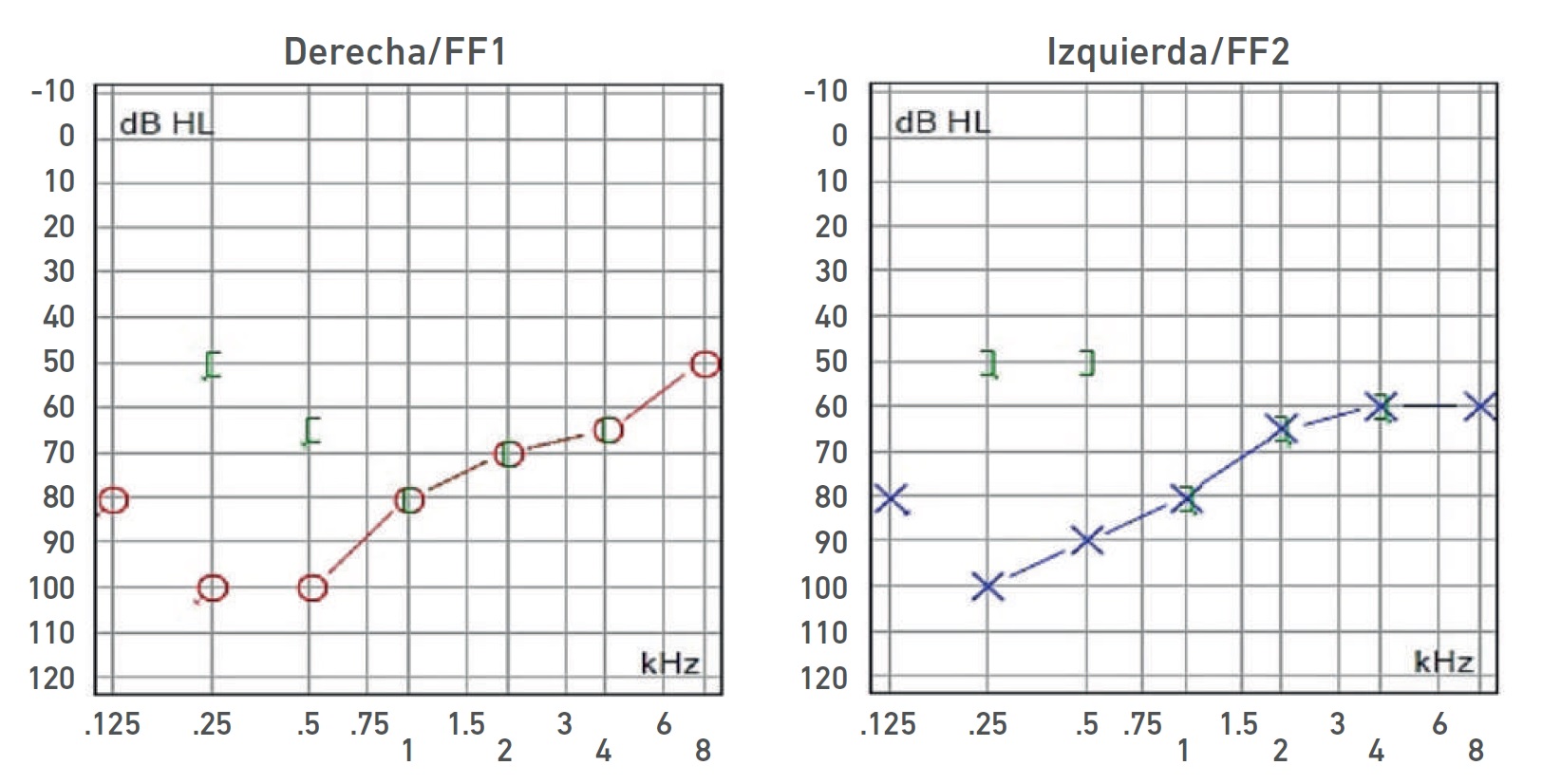
| IV-6 | HNS severa bilateral con curva de perfil ascendente, congénita | SIN TC | Imagen 6 |
| III-2 | Audición normal. Nota: madre portadora obligada | Normal |
Abreviaturas: Dx.: diagnóstico; MOI: Malformación de Oído interno; HNS: Hipoacusia Neurosensorial; PI- III: partición incompleta de cóclea Tipo III; PI - I: partición incompleta de cóclea Tipo I.
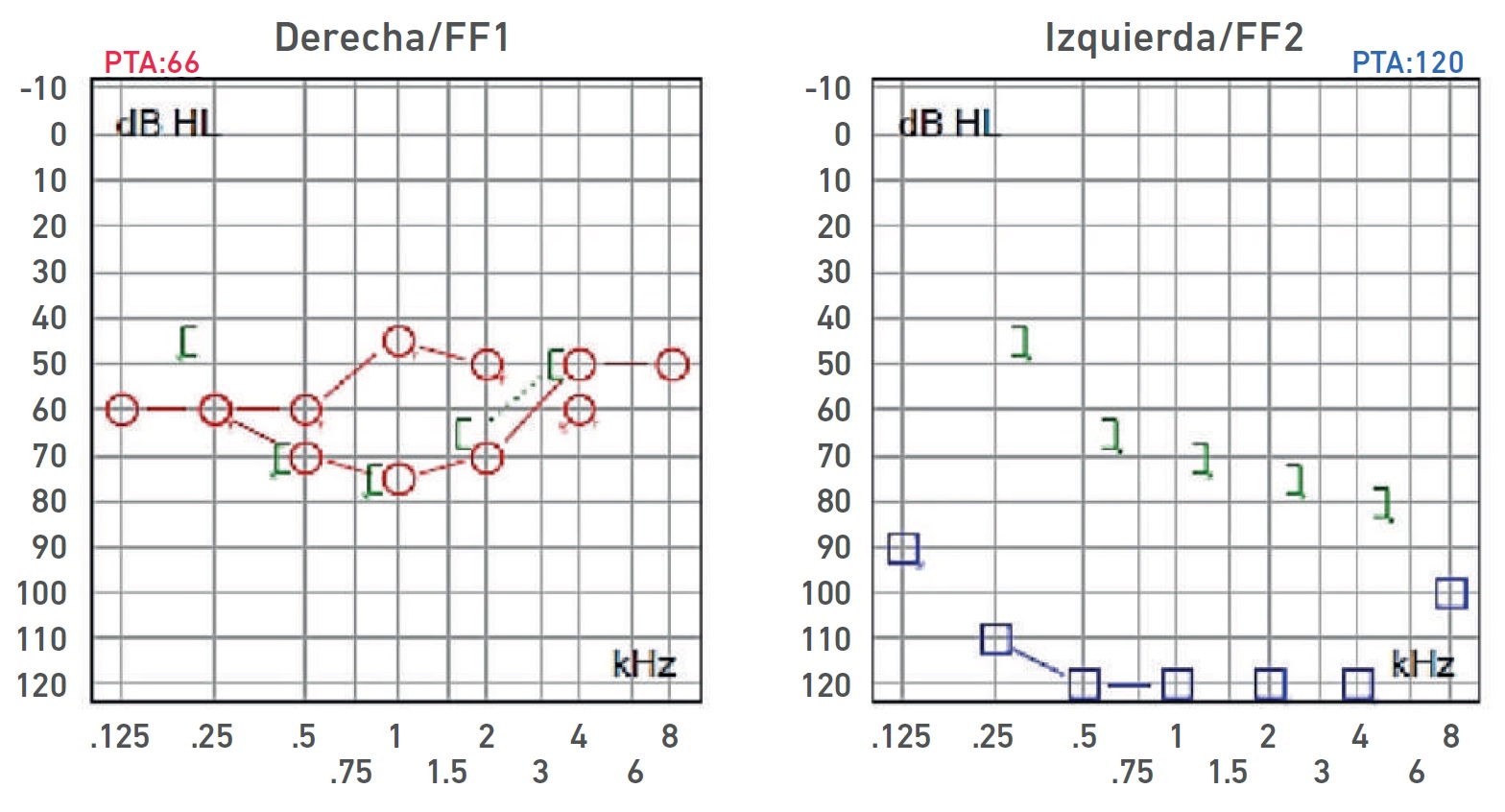
Nota. Audiometría: Oído derecho hipoacusia severa de tipo sensorial, curva de perfil irregular; oído izquierdo HNS con restos auditivos. Ganancia de auxiliar auditivo derecho con respuesta en promedio a 55 dB, sin llegar al área de lenguaje.
Imagen 3 Audiometría del propósito IV-1
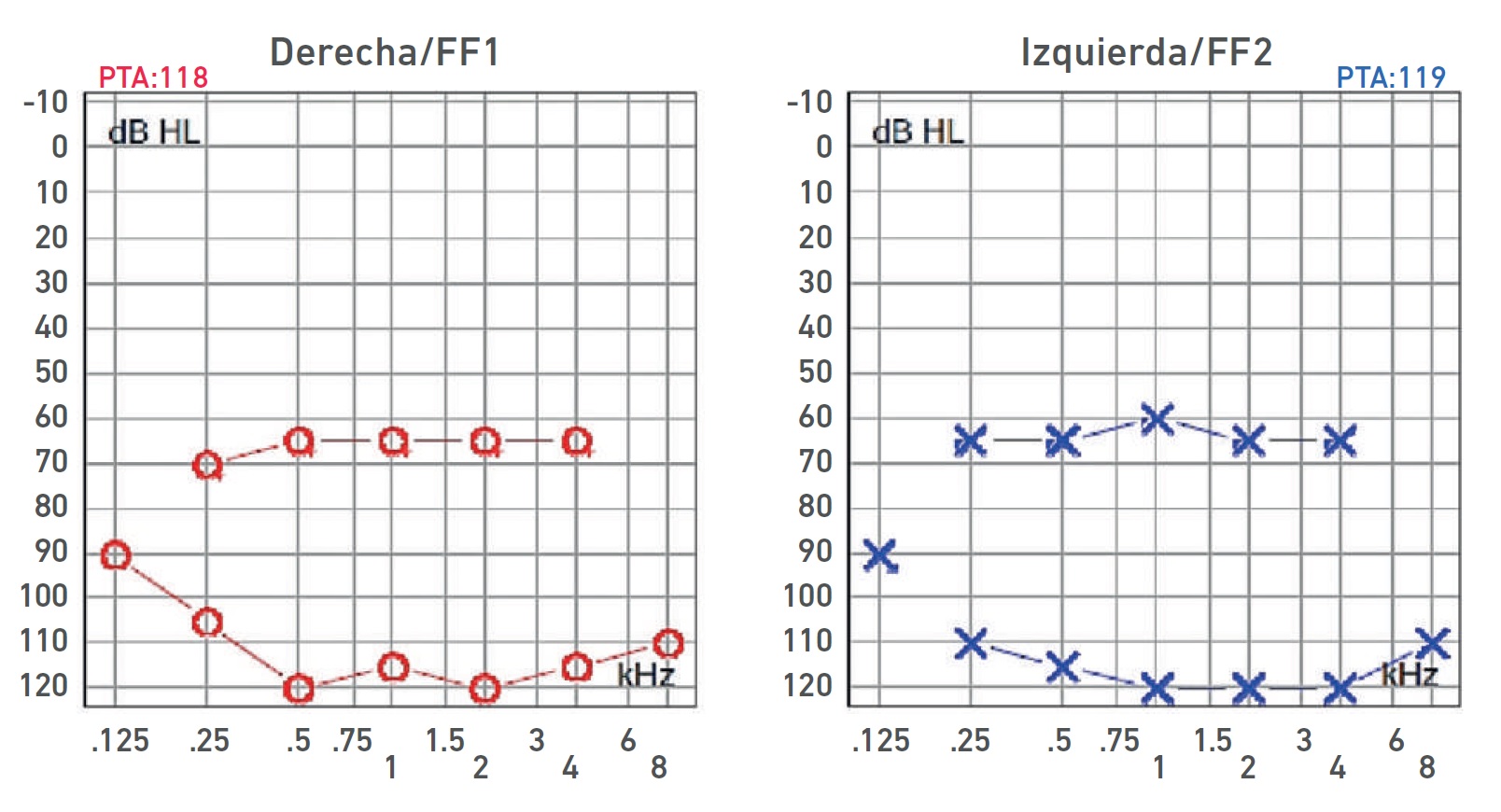
Nota. Audiometría: HNS profunda bilateral con curvas de perfil plano. Ganancia de auxiliares auditivos con respuesta en promedio a 65 dB de forma bilateral, sin llegar al área de lenguaje.
Imagen 4 Audiometría del paciente IV-2
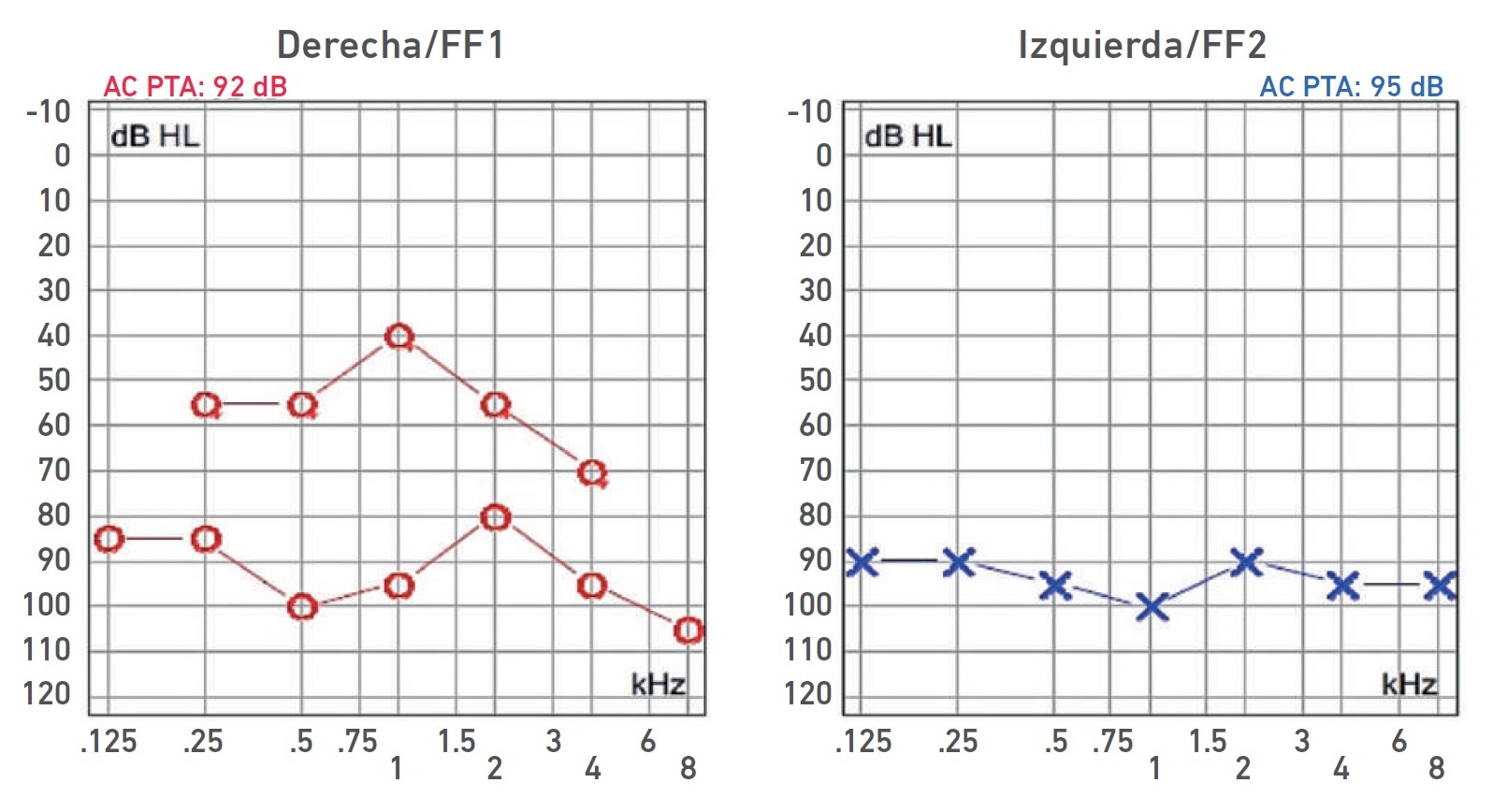
Nota. Audiometría: HNS profunda bilateral con curva de perfil irregular derecha y curva plana izquierda. Ganancia de auxiliar auditivo derecho con respuesta en promedio a 55 dB, sin llegar al área de lenguaje.
Imagen 5 Audiometría del paciente IV-4.
Nota. Audiometría: HNS severa bilateral con curva de perfil ascendente.
Imagen 6 Audiometría del paciente IV-6.
Severidad: Severa a profunda (solo en IV-3 fue de media a severa).
Lateralidad o simetría: bilateral.
Edad de aparición: Congénita.
Progresión: No.
Tipo de curva en el audiograma: variable dependiendo del grado de la MOI.
A cuatro individuos varones IV-5, III-5, II-1 y II-3, sordos, sin desarrollo de lenguaje, no se les realizó estudio audiológico, ni de imagen TC, por lo cual el diagnóstico clínico presuntivo es HNS de severa a profunda bilateral congénita, sordera clínica.
Los estudios audiológicos describen el fenotipo audiológico en nueve propósitos, como HNS de severa a profunda, bilateral, congénita. Este fue homogéneo en todos los afectados, salvo la variación de las curvas en la audiometría y en el paciente IV-3, cuyo grado de severidad fue de media a severa, como se observa en la Tabla 2, y quien tendrá que reevaluarse por audiometría lúdica cuando tenga la edad para cooperar.
Evaluación de Imagen (TC)
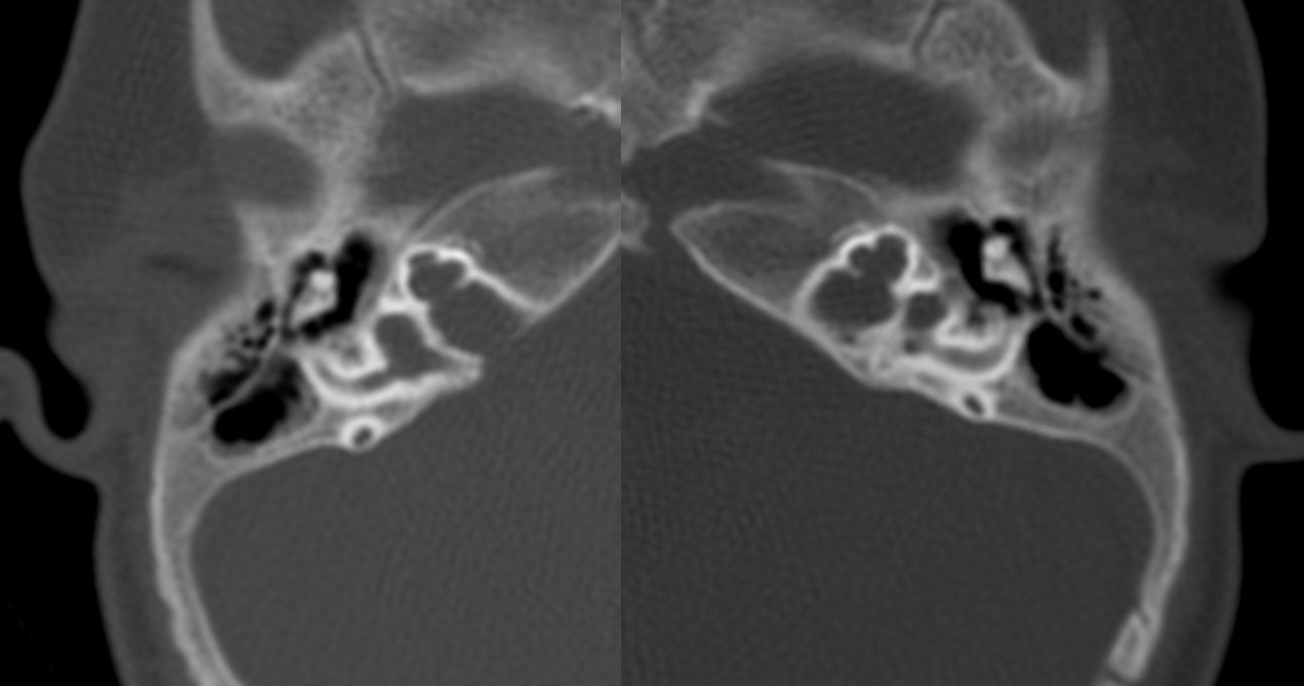
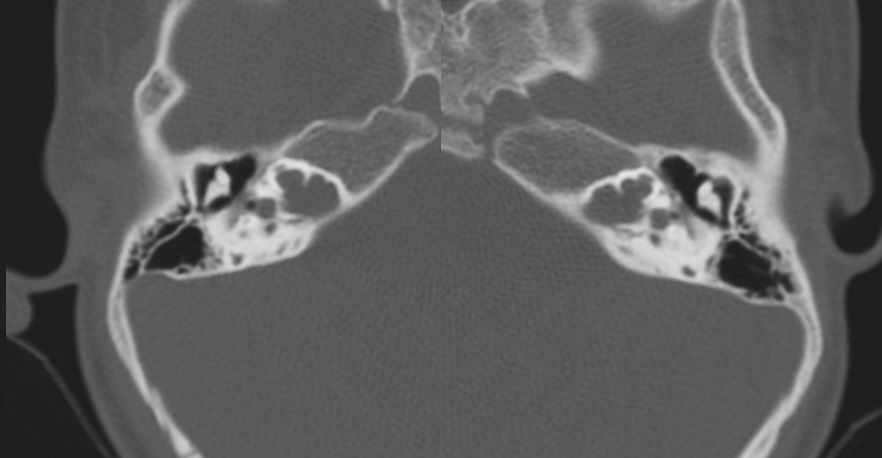
Los hallazgos radiológicos, mediante TC de oídos a cuatro pacientes varones y una madre oyente, portadora obligada, se analizaron y clasificaron según Sennanoğlu et al. [8,9]. En ese sentido, son consistentes con PI-III en tres de ellos: IV-1, IV -2 y IV- 4. El paciente IV-3 mostró PI-I; la madre portadora obligada no mostró alteración alguna (Ver Tabla 3). En los cuatro pacientes estudiados de esta familia observamos dos tipos de MOI, PI-I y PI-III, incluso entre los hermanos IV-2 y IV-3, que presentaron diferente tipo de PI, lo que evidencia expresividad variable. La malformación PI-III es de las MOI menos frecuente y se encuentra presente en este estudio familiar como predominante.
Tabla 3 Hallazgos de MOI, de expresividad variable, en cuatro propósitos, clasificados según Sennanoğlu et al. (8, 9, 13)
| Individuo | Cóclea | Vestíbulo | Otros | Clasificación | Ver |
|---|---|---|---|---|---|
| IV-1 | Ausencia de modiolo con presencia bilateral de septo interescalar | Discretamente amplio | Dilatación parcial en su segmento lateral | PI-III | Imagen 7 |
| IV-2 | De tipo quístico con ausencia bilateral de modiolo | Dilatado con hipoplasia de CSL izquierdo | Aparente estenosis de la entrada del nervio coclear al miodolo | PI-III | Imagen 8 |
| IV-3 | Ausencia de modiolo con presencia bilateral de septo interescalar | Dilatación en su segmento lateral | PI-I | Imagen 9 | |
| IV-4 | Ausencia de modiolo con presencia de septo interescalar bilateral | Discretamente amplio | Dilatación en su segmento lateral | PI-III | Imagen 10 |
| III-2 | Normal | Normal | Normal |
Abreviaturas: MOI: Malformación de oído interno; PI-III: partición incompleta de cóclea Tipo III; CSL: canal semicircular lateral; PI-I: partición incompleta de cóclea Tipo I.
Resultados Estudio Genético
Se muestra la genealogía de una familia mexicana con catorce individuos, nueve varones afectados con HNS de severa a profunda bilateral congénita y cinco madres portadoras obligadas oyentes. En las tres generaciones se observaron hermanos de los afectados de ambos sexos, oyentes, no representados en el árbol genealógico (AG) (Imagen 11). Todos fueron revisados e interrogados. No se encontraron datos fenotípicos que nos indicaran entidad sindrómica, por lo que se consideró hipoacusia no sindrómica. La Tabla 4 resume los diagnósticos clínicos de los individuos estudiados, representados en el AG.
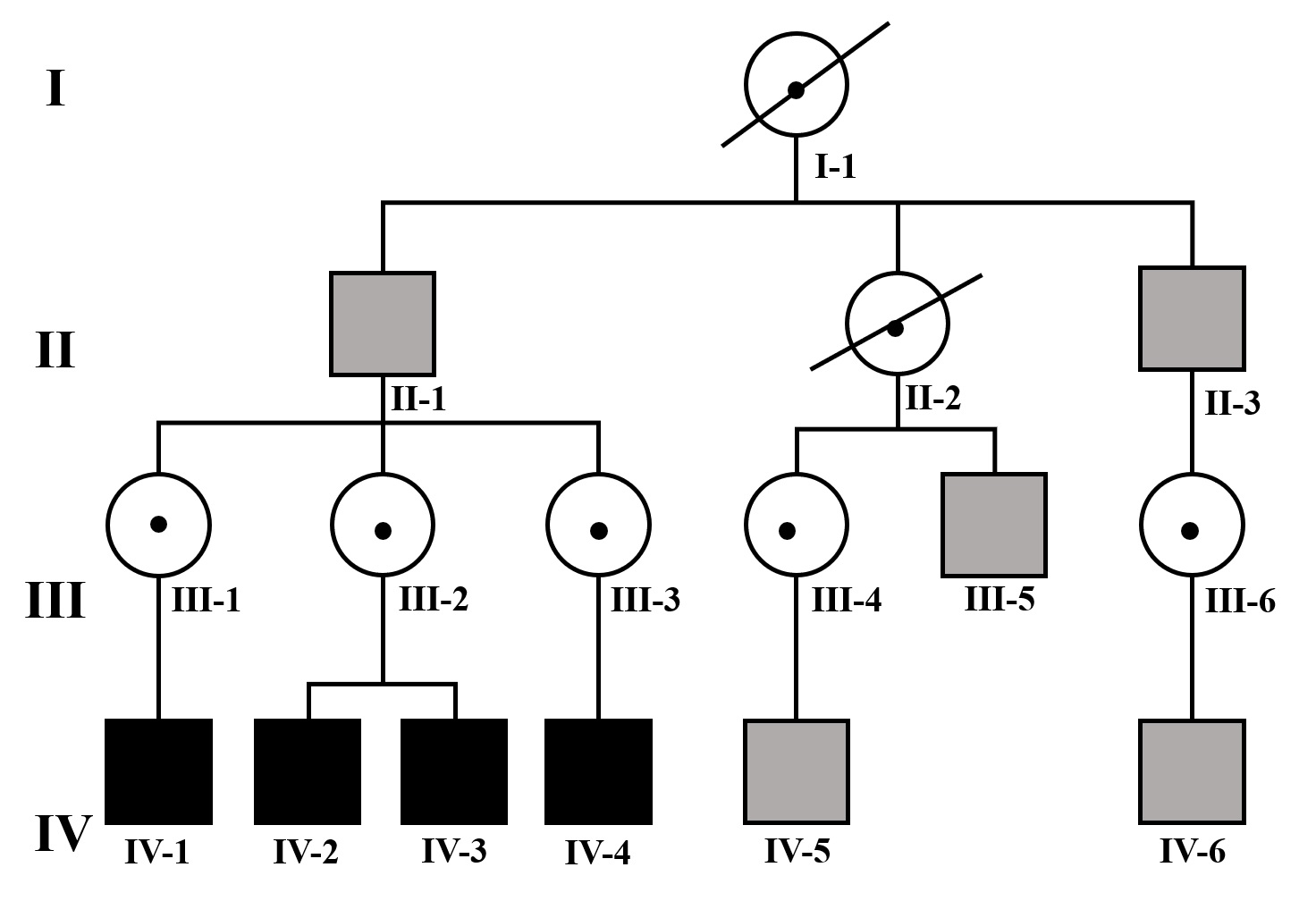
IV-1. PI-III. HNS severa derecha con curva de perfil irregular, HNS profunda izquierda, con restos auditivos, congénita.
IV-2. PI-III. HNS profunda bilateral con curvas de perfil plano, congénita.
IV-3. PI-I. HNS media bilateral, congénita.
IV-4. PI-III. HNS profunda bilateral, con curva de perfil irregular derecha y plana izquierda, congénita.
IV-6. HNS severa bilateral con curva de perfil ascendente, congénita.
I-1, II-2, III-1, III-2, III-4, III-6. Portadoras LXR.
II-1, II-3, III-5, IV5. HNS, profunda, bilateral, congénita (sordera clínica sin estudios).
Tabla 4 Resumen de Diagnóstico clínico de 14 individuos estudiados.
| Individuo | Dx. Audiológico | Dx. MOI | Imagen |
|---|---|---|---|
| IV-1 | HNS severa derecha con curva de perfil irregular. HNS profunda izquierda con restos auditivos, congénita | PI-III | 3 y 7 |
| IV-2 | HNS profunda bilateral con curvas de perfil plano, congénita | PI-III | 4 y 8 |
| IV-3 | HNS media bilateral congénita | PI-I | 1, 2 y 9 |
| IV-4 | HNS profunda bilateral con curva de perfil irregular derecha y curva plana izquierda, congénita | PI-III | 5 y 10 |
| IV-5 | HNS de severa a profunda, bilateral, congénita. Clínicamente Sordo, sin desarrollo de lenguaje | ||
| IV-6 | HNS severa bilateral con curva de perfil ascendente, congénita | 6 | |
| III-1 | Audición normal clínicamente, madre portadora | ||
| III-2 | Audición normal, madre portadora | Normal | |
| III-3 | Audición normal clínicamente, madre portadora | ||
| III-4 | Audición normal clínicamente, madre portadora | ||
| III-5 | HNS de severa a profunda, bilateral, congénita. Clínicamente Sordo, sin desarrollo de lenguaje | ||
| III-6 | Audición normal clínicamente, madre portadora | ||
| II-1 | HNS de severa a profunda, bilateral, congénita. Clínicamente Sordo, sin desarrollo de lenguaje | ||
| II-3 | HNS de severa a profunda, bilateral, congénita. Clínicamente Sordo, sin desarrollo de lenguaje |
Abreviaturas: MOI: Malformación de Oído interno; LXR: ligada a X recesiva; Dx.: Diagnostico; HNS: Hipoacusia Neurosensorial; PI-III: partición incompleta de cóclea Tipo III; PI-I: partición incompleta de cóclea Tipo I.
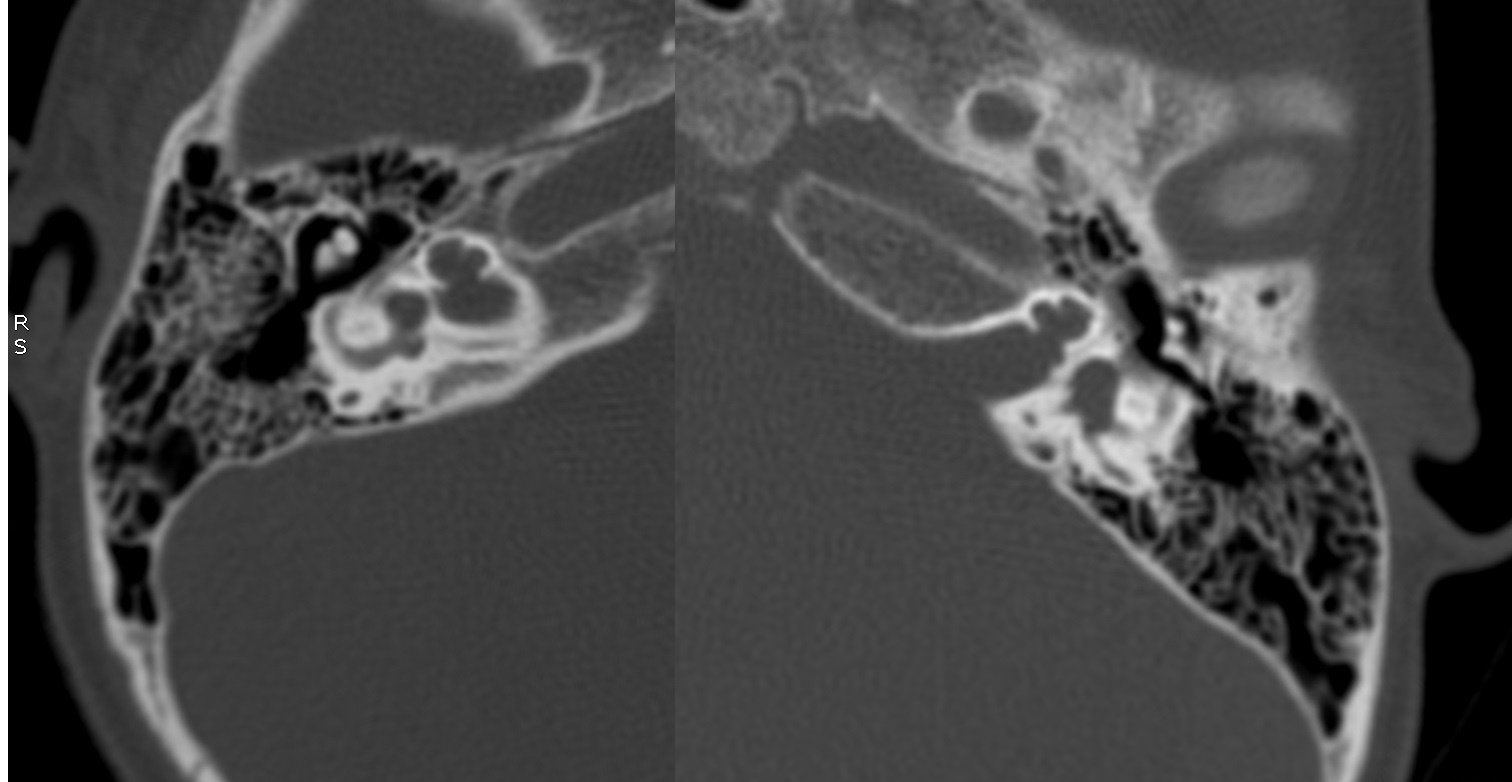
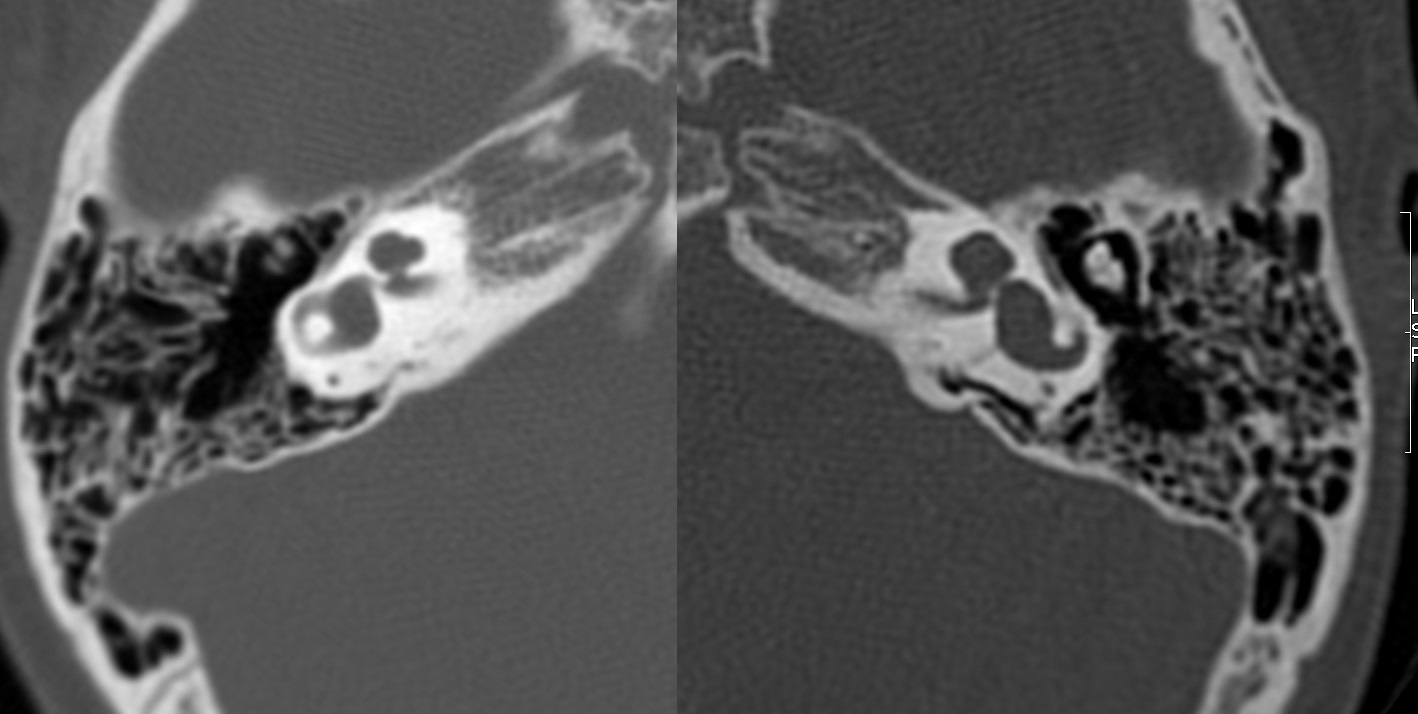
Las características clínico-genéticas de esta familia mexicana describen el fenotipo como HNS de severa a profunda bilateral congénita, con MOI tipo partición coclear incompleta (PI), de expresividad variable, de herencia LXR, no sindrómica.
Se creó banco de ADN de dieciséis miembros de la familia: ocho varones afectados, IV-1, IV-2, IV-3, IV-4, IV-5, IV-6, II-1 y II-3; cinco mujeres oyentes, madres portadoras obligadas III-1, III-2, III-3, III-4 y III-6; y tres hermanas de afectados, oyentes, posibles portadoras. Estos se enviaron al Dr. Mustafa Tekin del John P. Hussmann Institute for Human Genomics, Department of Human Genetics, Miami, USA para su estudio molecular, donde los resultados del estudio molecular de secuenciación de exoma completo (WES) están en proceso.
Discusión
La presencia de HNS congénita o prelingual, LX, se ha reportado en DFNX1, DFNX2, DFNX3 y DFNX6 [1,2]. Sin embargo, en DFNX1 es más frecuente que sea postlingual y sin MOI. En la DFNX2 es la única en la que se ha informado la presencia de PI-III y el tipo de la hipoacusia más frecuentemente encontrado es la mixta, por presentar fijación de la platina del estribo. [5,11,12]. La DFNX3 no presenta MOI y en la DFNX6 el grado de la hipoacusia es variable y no presenta PI-III [1,2]. En nuestros pacientes, cuyo árbol genealógico indica herencia ligada al X, y que sí presentaron MOI del tipo PI-PIII, es más probable que se trate de DFNX2, aunque no se encontró la fijación de la platina del estribo y el grado de la hipoacusia suele ser menor, pero el tipo DFNX1 que sí se acompaña de sordera neurosensorial, de severa a profunda y bilateral, no suele haber MOI y su presentación no es congénita [1,2,5,11,12,17].
Los pacientes estudiados en esta extensa familia muestran MOI que discrepan entre ellos y no es posible determinar, con los hallazgos obtenidos y las descripciones en la literatura, si pueden corresponder a alguno de los tipos descritos. Es probable que con el estudio molecular se pueda saber si forman parte de alguno de los tipos ya conocidos en los que existe un gen identificado, considerando expresividad variable, o que se trate de algún nuevo tipo, ya sea por ser gen diferente a los descritos o variantes diferentes de los ya descritos (Tabla 1) [5,11,12].
El fenotipo audiológico HNS de severa a profunda bilateral congénita, en los nueve varones afectados, fue homogéneo en tipo, severidad, lateralidad y presentación, con excepción del paciente de un año de edad (IV-3), evaluado por PEATC y por PEAEE, cuyos hallazgos mostraron HNS media a severa, congénita, diferente severidad a los otros ocho individuos afectados, pero este diagnóstico tendrá que corroborarse, por audiometría lúdica, cuando el individuo tenga edad para cooperar. Las curvas audiométricas mostraron variación entre todos los afectados estudiados, dependiente de los grados de la MOI.
Como ha sido descrito, algunas sorderas LX presentan progresión y en algunas portadoras se ha descrito pérdida auditiva de leve a media postlingual, de presentación tardía. En las mujeres, que de acuerdo al árbol genealógico son portadoras obligadas o probables, es necesario realizar seguimiento estrecho para conocer el comportamiento audiológico, porque no conocemos cómo será la evolución, ya que aparentemente pudiera ser un nuevo tipo de hipoacusia ligada al X.
El estudio con TC de una de las madres portadora no mostró MOI, por lo que supondríamos, que al menos en su caso, no existe expresividad menor del gen involucrado. Como se trata de una sola mujer portadora, no podemos asegurarlo, pero es probable que las portadoras no presenten manifestaciones menores, al menos en algunos casos. Es necesario, además del seguimiento audiológico, extender el estudio de imagen al resto de las portadoras.
Pensando en la posibilidad de realizar estudio molecular dirigido por el fenotipo se consideraron, de acuerdo al fenotipo, en dos posibilidades: el gen POU3F4 relacionado con DFNX2, como se mencionó, y en el SLC26A4, por ser el segundo más frecuentemente asociado con amplio espectro de MOI [5,7,11,12].
Consideramos que, fenotípicamente, los pacientes aquí descritos no parecen corresponder a ninguno de los siete tipos de DFNX descritos previamente y que es muy probable que algún otro gen sea el responsable del fenotipo descrito. Es necesario, por lo mismo, realizar estudio molecular amplio que incluya diversos genes y no solo los relacionados con DNFNX ya descritos.
La malformación coclear, tipo PI-III, observada en tres de los pacientes, es una de las MOI menos frecuentes; también, entre los grupos de PI [11] con patrón de herencia ligado al X y de la que no se han descrito, hasta donde sabemos, otros casos en México, consideramos que es importante informar al respecto en la literatura médica.
La fortaleza más importante del presente informe es haber realizado un estudio audiológico completo y tomográfico de algunos pacientes y una madre portadora, pero el hecho de que no se pudieran estudiar a todos los involucrados es también su debilidad.
Conclusiones
Se define el fenotipo de un tipo de HNS ligada al X.
Es probable que se trate de un tipo nuevo, ya que, por las características clínicas y paraclínicas presentadas por los pacientes, no se pudo encontrar coincidencia con alguno de los siete tipos de sorderas DFNX ya descritos.
Conocer el patrón de herencia nos permite llevar a cabo el adecuado asesoramiento genético en esta familia, que incluye la definición de los riesgos de transmisión, pero también la evolución y pronóstico, así como guiar la terapéutica a seguir.
Se contribuye a ampliar el conocimiento de las diferentes MOI con patrón de herencia ligada al X.
Se debe realizar en estos pacientes un exoma completo (WES en proceso) para determinar, en la medida de lo posible por las limitaciones del estudio, si existe alguna variante patogénica no descrita, que pueda ser la causante del fenotipo presente en los pacientes.

